Advanced Synthetic Route for High-Purity Flavonoid Intermediates and Commercial Scale-Up
The pharmaceutical industry is constantly seeking reliable sources for complex bioactive molecules, particularly those with demonstrated efficacy in treating cardiovascular conditions. Patent CN101712705B introduces a groundbreaking synthetic methodology for producing apigenin-7-O-β-D-glucopyranosyl-4'-O-α-L-rhamnopyranosid, also known as Formula A. This flavonoid compound has shown significant potential in treating ischemic diseases, including coronary heart disease and cerebral infarction, by reducing heart rate and myocardial contractility. Unlike traditional methods that rely on the inefficient extraction from Ranunculus plants, this patent outlines a robust chemical synthesis pathway that ensures high purity and scalability. For R&D directors and procurement managers, understanding this technology is crucial for securing a stable supply of high-purity pharmaceutical intermediates. The process leverages advanced organic synthesis techniques, including phase-transfer catalysis and Mitsunobu coupling, to construct the complex glycosidic bonds with precision. This shift from biological extraction to chemical synthesis represents a paradigm shift in how we approach the manufacturing of rare flavonoid derivatives, offering a solution to the supply bottlenecks that have historically plagued this sector.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the acquisition of Formula A has been hindered by its scarcity in natural sources. The background art highlights that extracting this compound from plants like Ranunculus sieboldii is incredibly inefficient, yielding merely 165 mg from 17 kg of dry plant material. This abysmal recovery rate makes it impossible to meet the demands of clinical research or commercial drug production. Furthermore, plant extraction processes are inherently variable, subject to seasonal changes, geographical differences, and environmental factors that affect the phytochemical profile of the source material. For a procurement manager, relying on such a volatile supply chain poses significant risks to project timelines and budget forecasting. The purification of the crude extract is also labor-intensive and often requires extensive chromatography to remove structurally similar impurities, driving up the cost per gram exponentially. These limitations render the natural extraction route commercially unviable for any application requiring kilogram or ton-scale quantities, effectively stalling the development of potential ischemic disease therapies.
The Novel Approach
The synthetic route disclosed in CN101712705B offers a transformative alternative by constructing the molecule from readily available starting materials through a series of controlled chemical reactions. Instead of hunting for trace amounts in nature, this method builds the flavonoid backbone and attaches the sugar moieties step-by-step. The core innovation lies in the use of specific acyl protecting groups, such as hexanoyl or benzoyl chains, which modulate the solubility and reactivity of the intermediates. This strategic protection allows for highly regioselective glycosylation, ensuring that the glucose and rhamnose units attach at the correct positions (7-O and 4'-O) without forming unwanted isomers. By utilizing phase-transfer catalysis in a heterogeneous solvent system, the reaction proceeds under mild conditions, avoiding the harsh reagents that often degrade sensitive flavonoid structures. This approach not only drastically improves the overall yield but also simplifies the purification process, making it feasible for cost reduction in pharmaceutical intermediate manufacturing on an industrial scale.
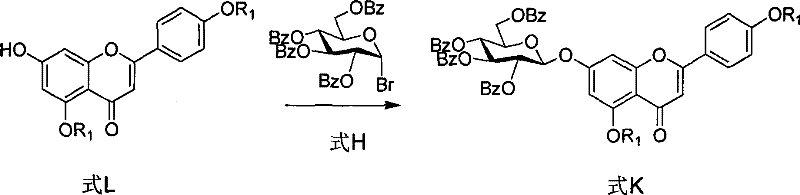
Mechanistic Insights into Phase-Transfer Catalyzed Glycosylation
The heart of this synthesis is the glycosylation step where the protected apigenin derivative (Formula L) reacts with 2,3,4,6-tetrabenzoyloxy bromoglucose (Formula H). This transformation is facilitated by a phase-transfer catalyst, typically tetrabutylammonium bromide, in a biphasic system of dichloromethane and water. The mechanism involves the generation of a phenoxide anion from the 7-hydroxyl group of Formula L by the action of a base like potassium carbonate. The phase-transfer catalyst shuttles this anion into the organic phase, where it attacks the anomeric carbon of the glucosyl bromide. This SN2-type displacement occurs with inversion of configuration, ensuring the formation of the desired β-glycosidic linkage. The use of benzoyl protecting groups on the glucose donor is critical; they provide steric bulk that favors the formation of the 1,2-trans glycosidic bond and prevent side reactions at other hydroxyl positions. For R&D teams, optimizing the molar ratio of the reactants (typically 1.5:1 of donor to acceptor) and the stirring efficiency is key to maximizing conversion while minimizing the formation of orthoesters or hydrolysis byproducts.
Following the initial glucosylation, the synthesis proceeds to attach the rhamnose unit via a Mitsunobu reaction. This step couples the 4'-hydroxyl group of the intermediate (Formula J) with 2,3,4-tribenzoyloxyrhamnose (Formula I). The Mitsunobu conditions, employing triphenylphosphine and diethyl azodicarboxylate (DEAD), activate the alcohol for nucleophilic substitution. This reaction is particularly valuable because it proceeds with inversion of stereochemistry at the anomeric center of the rhamnose, allowing the use of easily accessible precursors to generate the specific α-linkage required in Formula A. The careful control of temperature, typically between -20°C and 20°C, is essential to suppress side reactions and ensure high stereoselectivity. The final step involves a global deprotection using sodium methoxide in methanol, which cleanly removes all ester protecting groups to reveal the free hydroxyls of the target flavonoid glycoside. This mechanistic elegance ensures that the final product meets stringent purity specifications required for pharmaceutical applications.
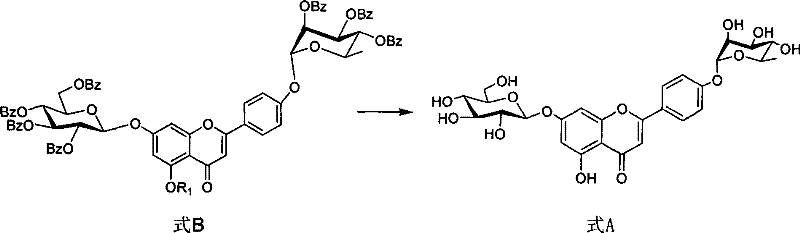
How to Synthesize Apigenin Glycoside Intermediates Efficiently
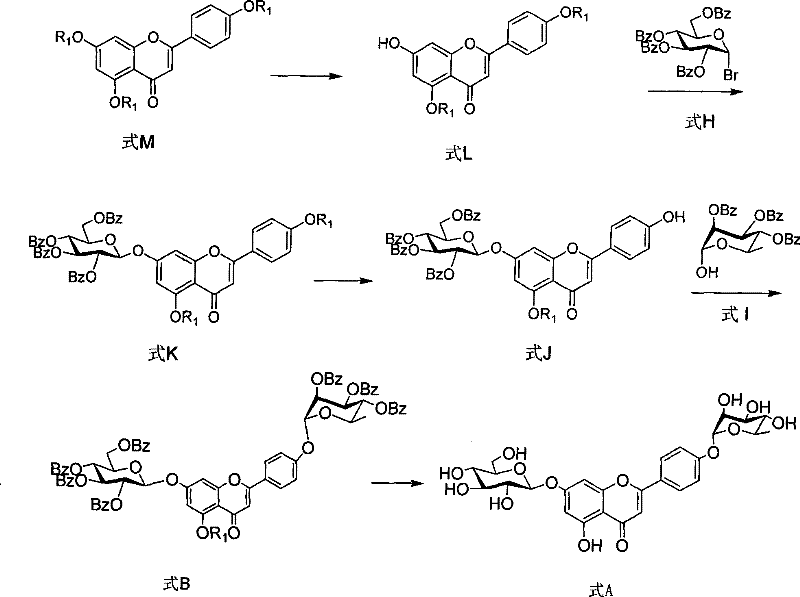
The synthesis of these complex flavonoid intermediates requires precise control over reaction parameters to ensure reproducibility and high yield. The process begins with the preparation of the aglycone, followed by sequential glycosylations and a final deprotection sequence. Each step has been optimized in the patent examples to balance reaction rate with selectivity. For instance, the selective deprotection of the 5-position acyl group while retaining the 7-position protection is a delicate operation that relies on the specific electronic and steric environment created by the hexanoyl chains. Operators must monitor the reaction progress closely using TLC to prevent over-deprotection. The detailed standardized synthesis steps见下方的指南 outline the exact stoichiometry, solvent volumes, and workup procedures necessary to achieve the reported yields of up to 80% for key intermediates. Adhering to these protocols is vital for maintaining the structural integrity of the flavonoid core throughout the multi-step sequence.
- Prepare the protected apigenin intermediate (Formula M) via acylation of apigenin with acyl chlorides.
- Perform selective deprotection to obtain Formula L, followed by phase-transfer glycosylation with tetrabenzoyl bromoglucose to yield Formula K.
- Execute Mitsunobu coupling between Formula J and tribenzoyl rhamnose (Formula I), followed by global deprotection to afford the final product Formula A.
Commercial Advantages for Procurement and Supply Chain Teams
For supply chain heads and procurement managers, the transition from plant extraction to total synthesis offers profound strategic advantages. The primary benefit is the decoupling of production from agricultural constraints. By synthesizing Formula A chemically, manufacturers are no longer at the mercy of crop yields, weather patterns, or geopolitical issues affecting the sourcing of rare medicinal plants. This shift ensures a consistent and predictable supply of the active ingredient, which is critical for maintaining continuous drug manufacturing lines. Furthermore, the synthetic route utilizes commodity chemicals and standard reagents that are readily available in the global market, reducing the risk of raw material shortages. The scalability of the process means that production can be ramped up from laboratory grams to commercial tons without fundamental changes to the chemistry, providing the flexibility needed to respond to market demand fluctuations.
- Cost Reduction in Manufacturing: The synthetic pathway eliminates the massive waste associated with processing tons of plant biomass to recover milligrams of product. By starting with inexpensive apigenin and simple acyl chlorides, the material cost basis is significantly lowered. Additionally, the use of phase-transfer catalysis allows reactions to proceed at moderate temperatures (30°C to 50°C), reducing energy consumption compared to high-temperature reflux processes. The high selectivity of the reactions minimizes the formation of difficult-to-remove impurities, thereby reducing the cost and time associated with downstream purification and chromatography. These efficiencies collectively contribute to substantial cost savings in pharmaceutical intermediate manufacturing, making the final therapeutic more economically viable.
- Enhanced Supply Chain Reliability: Relying on a chemical synthesis route diversifies the supply chain and mitigates the risks inherent in botanical sourcing. The intermediates, such as Formula K and Formula J, are stable solids that can be stockpiled, acting as a buffer against supply disruptions. The robustness of the chemistry means that production can be transferred between different manufacturing sites with minimal technology transfer friction, ensuring business continuity. For procurement teams, this translates to stronger negotiating power and the ability to secure long-term contracts with reliable flavonoid intermediates suppliers who can guarantee delivery schedules regardless of external environmental factors.
- Scalability and Environmental Compliance: The process is designed with green chemistry principles in mind, utilizing solvents like dichloromethane and methanol which can be recovered and recycled efficiently. The avoidance of heavy metal catalysts simplifies waste treatment and ensures compliance with strict environmental regulations regarding residual metals in pharmaceutical products. The high atom economy of the glycosylation and coupling steps reduces the volume of chemical waste generated per kilogram of product. This environmental efficiency not only lowers disposal costs but also aligns with the sustainability goals of modern pharmaceutical companies, facilitating easier regulatory approval and market acceptance for the final drug product.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of these flavonoid compounds. Understanding these details helps stakeholders evaluate the feasibility of integrating this technology into their existing pipelines. The answers are derived directly from the experimental data and claims within the patent documentation, ensuring accuracy and relevance for technical decision-makers.
Q: What is the primary advantage of this synthetic method over plant extraction?
A: The synthetic method described in CN101712705B overcomes the extremely low natural abundance of Formula A in plants (only 165mg from 17kg of dry weight), enabling large-scale industrial production with consistent purity and yield.
Q: What are the critical reaction conditions for the glycosylation step?
A: The glycosylation of Formula L with Formula H utilizes a heterogeneous system of dichloromethane and water with potassium carbonate as the base and tetrabutylammonium bromide as the phase transfer catalyst at 30°C to 50°C.
Q: How is the regioselectivity controlled during the synthesis?
A: Regioselectivity is achieved through the use of specific protecting groups (hexanoyl or benzoyl) at the 5 and 4' positions, leaving the 7-hydroxyl group available for glucosylation and the 4'-hydroxyl for subsequent rhamnosylation.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Apigenin Glycoside Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of having a dependable partner for complex pharmaceutical intermediates. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. We understand that the synthesis of flavonoid glycosides requires meticulous attention to detail, particularly in controlling stereochemistry and purity. Our state-of-the-art rigorous QC labs and stringent purity specifications guarantee that every batch of intermediate we deliver meets the highest industry standards. Whether you are in the early stages of drug discovery or preparing for commercial launch, our infrastructure is designed to support your growth and mitigate supply chain risks effectively.
We invite you to collaborate with us to leverage this advanced synthetic technology for your projects. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact us to request specific COA data and route feasibility assessments that demonstrate how our manufacturing capabilities can optimize your production costs and timelines. By partnering with NINGBO INNO PHARMCHEM, you gain access to a reliable apigenin glycoside supplier committed to delivering excellence in quality and service, ensuring the success of your cardiovascular therapeutic developments.