Advanced Asymmetric Catalytic Synthesis of Stepholidine for Commercial Pharmaceutical Manufacturing
The pharmaceutical industry's demand for optically pure central nervous system (CNS) agents has driven significant innovation in asymmetric synthesis, particularly for complex alkaloids like Stepholidine (1-SPD). Patent CN101921229B introduces a groundbreaking chemical synthesis method for L-Stepholidine and its derivative optical isomers, addressing the critical limitations of natural extraction and traditional racemic resolution. This technology leverages chiral metal catalysts to establish stereochemistry with high precision, offering a viable pathway for the commercial manufacturing of high-purity pharmaceutical intermediates. Unlike previous methods that relied on the scarce natural resources of the Stephania plant, where content is often below 1‰, this synthetic approach ensures a stable, scalable supply chain essential for modern drug development. The core innovation lies in the asymmetric catalytic hydrogenation of 3,4-dihydroisoquinoline compounds, a strategy that fundamentally shifts the production paradigm from resource-dependent extraction to precise chemical engineering.
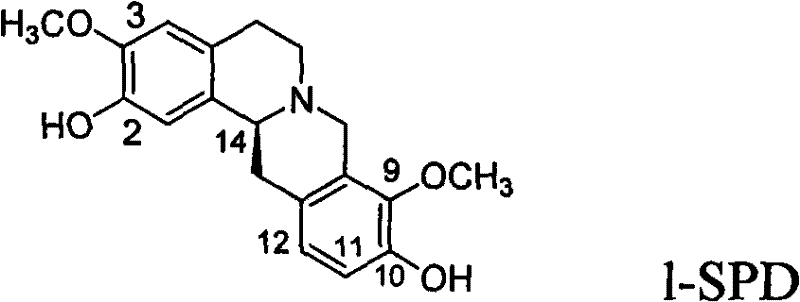
For R&D directors evaluating process feasibility, understanding the limitations of conventional methods is paramount. Historically, the production of 1-SPD relied heavily on isolation from dried roots and stems of Stephania plants, a process plagued by extremely low recovery rates and seasonal variability. Furthermore, early synthetic attempts produced racemic mixtures requiring diastereomeric resolution. Theoretical maximum yields for resolution processes cap at 50%, meaning half of the synthesized material is discarded as unwanted isomers, creating substantial chemical waste and inflating raw material costs. This inefficiency is unacceptable in modern green chemistry standards. In contrast, the novel approach detailed in the patent utilizes a chiral synthesis method that bypasses resolution entirely. By employing specific chiral metal catalysts during the hydrogenation step, the process directly generates the desired optical isomer. This not only potentially doubles the theoretical yield compared to resolution but also simplifies the purification workflow, reducing solvent consumption and downstream processing time significantly.
Mechanistic Insights into Ru-Catalyzed Asymmetric Transfer Hydrogenation
The heart of this synthetic breakthrough is the stereoselective reduction of the 3,4-dihydroisoquinoline intermediate (Formula I) to the tetrahydroisoquinoline derivative (Formula II). This transformation is mediated by chiral Ruthenium catalysts, such as those represented by formulas A1-A4 or B1-B4, typically involving TsDPEN ligands. The mechanism involves a transfer hydrogenation process where a hydrogen donor system, preferably a formic acid-triethylamine azeotrope (5:2 ratio), serves as the source of hydride and proton. The chiral environment created by the ligand around the Ruthenium center dictates the facial selectivity of the hydrogen addition to the imine double bond. This precise control allows for the establishment of the chiral carbon at the C14 position with exceptional enantiomeric excess, often reaching levels above 95% ee as demonstrated in the experimental examples. The reaction proceeds under mild conditions, typically between 0°C and room temperature, which minimizes side reactions and degradation of sensitive functional groups.
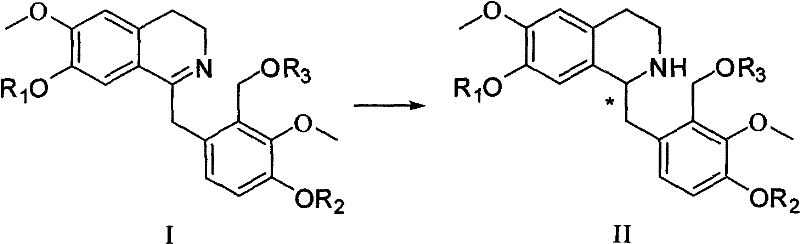
Following the critical hydrogenation step, the synthesis proceeds through a series of protective group manipulations and cyclizations designed to maintain optical integrity. The removal of the R3 protecting group (e.g., acetyl) reveals a free hydroxyl group, which is then subjected to intramolecular cyclization. This can be achieved via a Mitsunobu reaction using reagents like DIAD and triphenylphosphine, or alternatively through halogenation followed by base-mediated amination. These steps close the D-ring of the tetrahydroprotoberberine skeleton. Finally, the removal of phenolic protecting groups (R1 and R2), such as benzyl or isopropyl groups, yields the final active pharmaceutical ingredient. The entire sequence is designed to minimize racemization, ensuring that the high optical purity established in the early catalytic step is preserved through to the final product. This rigorous control over impurity profiles is crucial for meeting the stringent regulatory requirements for CNS drugs.
How to Synthesize Stepholidine Efficiently
The synthesis of Stepholidine via this patented route involves a convergent strategy starting from readily available substituted phenethylamines and lactone intermediates. The process integrates amide formation, cyclization, and the pivotal asymmetric hydrogenation step to construct the complex tetracyclic core. Detailed operational parameters, including specific solvent systems like anhydrous DMF for the hydrogenation and precise temperature controls for the Bischler-Napieralski reaction, are critical for success. For a comprehensive breakdown of the standardized operating procedures and safety protocols required for laboratory or pilot-scale execution, please refer to the technical guide below.
- Condensation of substituted phenethylamine with lactone intermediates followed by acylation and Bischler-Napieralski cyclization to form the 3,4-dihydroisoquinoline precursor.
- Execution of asymmetric catalytic hydrogenation using chiral Ruthenium-TsDPEN catalysts and a formic acid-triethylamine hydrogen donor system to establish the critical chiral center.
- Final deprotection and intramolecular cyclization via Mitsunobu reaction or halogenation-amination sequences to yield the target tetrahydroprotoberberine skeleton.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition from extraction to this asymmetric synthetic route represents a strategic opportunity to de-risk the supply of critical CNS intermediates. The traditional reliance on plant extraction exposes buyers to agricultural risks, including climate change impacts, harvest failures, and geopolitical instability in sourcing regions. By adopting this synthetic methodology, organizations can secure a consistent, year-round supply of high-purity Stepholidine that is immune to biological variability. Furthermore, the elimination of the resolution step inherently reduces the raw material burden. Since the process does not discard 50% of the product as the wrong enantiomer, the effective cost per kilogram of active material is drastically lowered, even if the catalyst costs are initially higher. This efficiency translates directly into improved margins and more competitive pricing for downstream drug formulations.
- Cost Reduction in Manufacturing: The primary economic driver of this technology is the atom economy gained by avoiding racemic resolution. In traditional resolution, more than half of the synthesized material is waste, requiring disposal and incurring environmental fees. This asymmetric route theoretically utilizes all starting material to form the desired isomer, significantly reducing the cost of goods sold (COGS). Additionally, the use of transfer hydrogenation eliminates the need for high-pressure hydrogenation reactors, which are capital-intensive and require specialized safety infrastructure. The ability to run reactions at atmospheric pressure with liquid hydrogen donors simplifies the equipment requirements, allowing for production in standard glass-lined or stainless steel reactors found in most multipurpose chemical facilities.
- Enhanced Supply Chain Reliability: Synthetic routes provide a level of predictability that natural extraction cannot match. The lead time for producing complex alkaloids via this method is determined by chemical reaction times and logistics rather than growing seasons. This reliability allows for better inventory planning and reduces the need for excessive safety stock. Moreover, the starting materials, such as substituted benzaldehydes and phenethylamines, are commodity chemicals available from multiple global suppliers, preventing single-source bottlenecks. This diversification of the raw material base ensures continuity of supply even if one vendor faces disruptions, a critical factor for maintaining uninterrupted pharmaceutical production lines.
- Scalability and Environmental Compliance: The process is designed with scale-up in mind, utilizing common organic solvents like ethanol, dichloromethane, and acetonitrile which have well-established recovery and recycling protocols. The reduction in chemical waste, specifically the absence of unwanted enantiomers, aligns with increasingly strict environmental regulations regarding pharmaceutical manufacturing effluents. The catalytic nature of the key step means that expensive chiral ligands are used in sub-stoichiometric amounts (often 0.1-2 mol%), further reducing the environmental footprint and cost. This combination of high efficiency and lower waste generation makes the process highly attractive for large-scale commercial manufacturing, facilitating the transition from gram-scale R&D to multi-ton annual production without significant process re-engineering.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this asymmetric synthesis technology. These insights are derived directly from the patent specifications and are intended to clarify the feasibility and benefits for potential partners. Understanding these details is essential for making informed decisions about integrating this route into your existing manufacturing portfolio.
Q: What is the primary advantage of this asymmetric synthesis method over traditional extraction?
A: Traditional extraction from plant sources yields less than 1% Stepholidine, making it unsustainable for large-scale demand. This synthetic route offers a scalable, consistent supply independent of agricultural variables, with theoretical yields far exceeding natural isolation limits.
Q: How does the chiral catalyst impact the enantiomeric purity of the final product?
A: The use of chiral Ruthenium metal catalysts (such as RuCl[(R,R)-TsDPEN(p-cymene)]) in the hydrogenation step allows for the direct establishment of the desired S or R configuration with high enantiomeric excess (ee), often exceeding 95%, eliminating the need for inefficient resolution steps.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the process utilizes robust reaction conditions, including transfer hydrogenation which avoids high-pressure hydrogen gas equipment, and standard organic solvents like dichloromethane and ethanol, facilitating safe and efficient scale-up from kilogram to multi-ton production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Stepholidine Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of asymmetric catalysis in the production of high-value CNS intermediates like Stepholidine. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory innovation to industrial reality is seamless. Our state-of-the-art facilities are equipped to handle the specific requirements of chiral synthesis, including moisture-sensitive operations and precise temperature control needed for high enantioselectivity. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch meets the highest international standards for pharmaceutical intermediates.
We invite you to collaborate with us to leverage this advanced synthetic technology for your supply chain. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this route can optimize your overall procurement budget. Contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us help you secure a sustainable, high-quality supply of Stepholidine for your next-generation antipsychotic therapies.