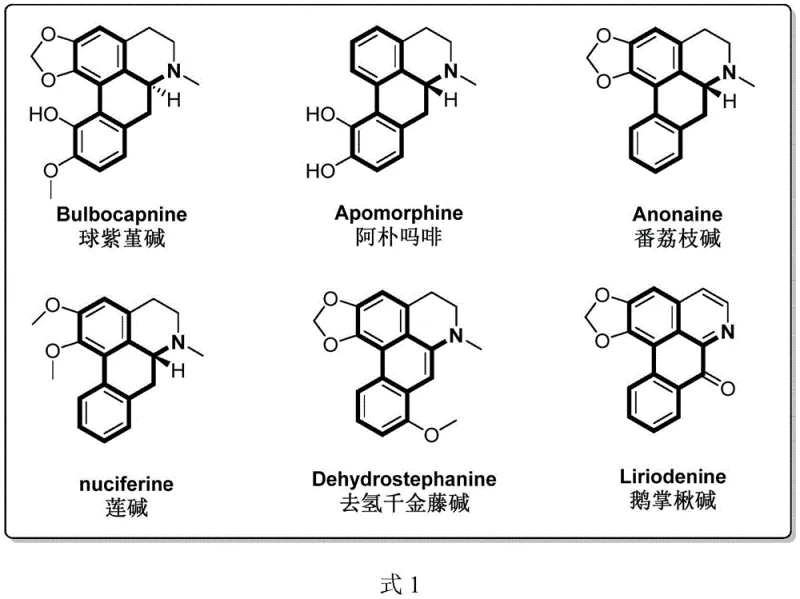
Advanced Synthesis of Aporphine Alkaloid Intermediates via Palladium Catalysis
Advanced Synthesis of Aporphine Alkaloid Intermediates via Palladium Catalysis
The pharmaceutical industry continuously seeks more efficient pathways to access complex natural product scaffolds, particularly those with significant therapeutic potential. Patent CN112239425B introduces a groundbreaking methodology for the preparation of apophylline alkaloid intermediates, leveraging advanced transition metal catalysis to streamline the construction of the tetracyclic core. This innovation addresses long-standing challenges in the synthesis of isoquinoline alkaloids, which are renowned for their diverse biological activities including anti-Parkinson's, anti-tumor, and antiviral properties. By shifting from traditional multi-step condensations to a direct C-H functionalization strategy, this technology offers a robust platform for generating high-value pharmaceutical intermediates. For R&D directors and procurement specialists, understanding the nuances of this patent is critical for securing a reliable pharmaceutical intermediate supplier capable of delivering complex scaffolds with improved cost-efficiency and supply continuity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the tetrahydrodibenzo[de,g]quinoline skeleton found in aporphine alkaloids has relied on laborious and chemically demanding sequences. Traditional routes typically commence with the condensation of aromatic ethylamines and aromatic acetic acids to form amide substrates, followed by a Bischler-Napieralski intramolecular cyclization to generate tetrahydroquinoline precursors. The critical bottleneck lies in the final ring-closing step, which often necessitates harsh conditions such as the classical Pschorr reaction, phenolic oxidative coupling, or radical cyclization. These legacy methods suffer from poor atom economy, limited substrate tolerance, and the requirement for specialized, often hazardous reagents. Furthermore, the sensitivity of these reactions to substituent patterns on the aromatic ring frequently leads to inconsistent yields and complex impurity profiles, complicating the purification process and driving up the cost of goods for the final API intermediate.
The Novel Approach
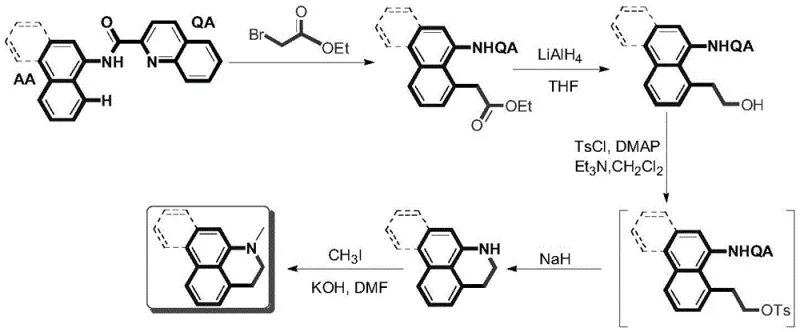
In stark contrast, the methodology disclosed in CN112239425B employs a modern palladium-catalyzed C-H activation strategy that fundamentally simplifies the synthetic logic. The process initiates with the direct alkylation of N-aryl-quinoline-2-carboxamide with ethyl bromoacetate, facilitated by a palladium catalyst and a potassium benzoate additive. This key transformation installs the necessary carbon side chain with high regioselectivity, bypassing the need for pre-functionalized halogenated starting materials. Subsequent steps involve a straightforward reduction of the ester moiety to an alcohol, followed by tosylation and a base-mediated intramolecular cyclization. This novel approach not only reduces the overall step count but also operates under milder conditions that are far more amenable to large-scale manufacturing. The versatility of this route allows for the introduction of various functional groups without compromising the ring construction, thereby enabling the synthesis of a broad library of aporphine derivatives from a common intermediate platform.
Mechanistic Insights into Pd-Catalyzed C-H Functionalization
The core innovation of this technology lies in the palladium-catalyzed ortho-C-H alkylation of the N-aryl-quinoline-2-carboxamide substrate. Mechanistically, the quinoline amide moiety acts as a directing group, coordinating the palladium center to the proximal C-H bond on the aryl ring. In the presence of the oxidant and the alkyl halide (ethyl bromoacetate), the catalyst facilitates the cleavage of the inert C-H bond and the formation of a new C-C bond. The inclusion of PhCOOK as an additive is crucial, likely serving to regenerate the active catalytic species or stabilize the transition state, thereby enhancing the turnover number of the expensive palladium catalyst. This directed metalation strategy ensures high regioselectivity, minimizing the formation of undesired isomers that are common in non-directed electrophilic aromatic substitutions. Following the alkylation, the ester group serves as a latent handle for ring closure; reduction with lithium aluminum hydride converts it to a primary alcohol, which is subsequently activated as a tosylate.
The final cyclization step is driven by the nucleophilicity of the amide nitrogen. Upon treatment with a strong base like sodium hydride, the deprotonated nitrogen attacks the electrophilic carbon bearing the tosylate group in an intramolecular SN2 reaction. This elegant cascade effectively constructs the fourth ring of the aporphine skeleton, yielding the 2,3-dihydro-1H-benzo[d]quinoline core. From an impurity control perspective, this sequence is highly advantageous. The distinct chemical nature of each intermediate allows for effective purification via standard silica gel chromatography or crystallization. Moreover, the mildness of the cyclization conditions prevents the degradation of sensitive functional groups that might be present on the aromatic rings, ensuring a clean impurity profile. This level of control is paramount for meeting the stringent quality specifications required for high-purity pharmaceutical intermediates intended for clinical applications.
How to Synthesize Aporphine Alkaloid Intermediates Efficiently
The synthesis of these valuable scaffolds is achieved through a logical sequence of transformations that balance reactivity with operational simplicity. The process begins with the preparation of the N-aryl-quinoline-2-carboxamide starting material, followed by the pivotal palladium-catalyzed alkylation. Detailed protocols for reaction temperatures, solvent choices, and workup procedures are essential to maximize yield and minimize waste. The subsequent reduction and cyclization steps utilize standard reagents found in most fine chemical facilities, facilitating easy technology transfer. For process chemists looking to implement this route, understanding the stoichiometry and safety parameters of reagents like LiAlH4 and NaH is critical. The detailed standardized synthesis steps see the guide below.
- Perform Pd-catalyzed ortho-alkylation of N-aryl-quinoline-2-carboxamide with ethyl bromoacetate using PhCOOK as an additive.
- Reduce the resulting ester intermediate to the corresponding alcohol using LiAlH4 in THF.
- Convert the alcohol to a tosylate leaving group, followed by base-mediated intramolecular cyclization with NaH to form the core ring system.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis route translates into tangible strategic benefits beyond mere technical elegance. The shift towards a C-H functionalization-based strategy significantly de-risks the supply chain by relying on broadly available commodity chemicals rather than bespoke, multi-step custom building blocks. This simplification of the bill of materials enhances supply security and reduces the vulnerability to disruptions in the sourcing of exotic reagents. Furthermore, the robustness of the reaction conditions means that the process is less prone to batch-to-batch variability, a common pain point in the manufacturing of complex heterocyclic compounds. By streamlining the synthesis, manufacturers can achieve faster turnaround times from order to delivery, directly addressing the industry-wide challenge of reducing lead time for high-purity pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The elimination of multiple protection-deprotection sequences and the use of catalytic rather than stoichiometric amounts of expensive metals drive down the raw material costs. Additionally, the higher overall yield resulting from fewer synthetic steps means less solvent consumption and reduced waste disposal costs. The process avoids the use of extremely low-temperature conditions or high-pressure equipment, allowing for production in standard glass-lined reactors, which further optimizes capital expenditure and operational expenses associated with cost reduction in API manufacturing.
- Enhanced Supply Chain Reliability: The starting materials, such as quinoline-2-carboxylic acid and various naphthylamines or phenanthrenylamines, are commercially accessible from multiple global vendors. This diversification of the supply base prevents single-source bottlenecks. The chemical stability of the intermediates allows for flexible inventory management, where key precursors can be stockpiled without significant degradation. This reliability is crucial for maintaining continuous production schedules for downstream drug substances, ensuring that partners have a reliable pharmaceutical intermediate supplier who can meet demand fluctuations without compromise.
- Scalability and Environmental Compliance: The reaction solvents employed, such as dichloroethane and THF, are well-understood in industrial settings with established recovery and recycling protocols. The avoidance of heavy metal waste streams typical of older coupling methods simplifies environmental compliance and wastewater treatment. The scalability of the Pd-catalyzed step has been demonstrated to maintain efficiency upon scale-up, supporting the commercial scale-up of complex pharmaceutical intermediates from pilot plant quantities to multi-ton annual production capacities required for blockbuster drug candidates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on the practical aspects of the method. Understanding these details helps stakeholders evaluate the feasibility of integrating this route into their existing supply chains.
Q: What are the key advantages of this Pd-catalyzed route over traditional methods?
A: This method utilizes direct C-H functionalization, eliminating the need for pre-functionalized starting materials and reducing the total number of synthetic steps compared to classical Bischler-Napieralski or Pschorr reactions.
Q: Is this synthesis scalable for commercial production?
A: Yes, the protocol uses commercially available reagents like palladium acetate and standard solvents (DCE, THF, DMF), making it highly suitable for scale-up from kilogram to multi-ton production.
Q: What is the purity profile of the final intermediates?
A: The process includes robust purification steps such as column chromatography and recrystallization, ensuring high-purity intermediates suitable for downstream API synthesis.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Aporphine Alkaloid Intermediate Supplier
The technological advancements detailed in CN112239425B represent a significant leap forward in the accessibility of bioactive aporphine scaffolds. NINGBO INNO PHARMCHEM stands at the forefront of translating such innovative academic and patent literature into commercial reality. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project moves seamlessly from gram-scale optimization to full-scale manufacturing. Our commitment to quality is underpinned by stringent purity specifications and rigorous QC labs equipped with state-of-the-art analytical instrumentation, guaranteeing that every batch of high-purity pharmaceutical intermediates meets the exacting standards of the global pharmaceutical industry.
We invite you to leverage our technical expertise to optimize your supply chain and reduce your overall cost of goods. By partnering with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact our technical procurement team today to request specific COA data for our catalog items or to discuss route feasibility assessments for your proprietary compounds. Let us collaborate to bring your next-generation therapeutics to market faster and more efficiently.