Scalable Synthesis of 2-Fluoro-3-Hydroxypyridine-4-Carboxylic Acid via Regioselective Lithiation
The pharmaceutical and agrochemical industries are constantly seeking efficient pathways to access fluorinated heterocycles, which serve as critical building blocks for next-generation therapeutics. Patent CN110669002A introduces a groundbreaking synthetic methodology for producing 2-fluoro-3-hydroxypyridine-4-carboxylic acid, a valuable intermediate characterized by its unique substitution pattern. This innovation addresses long-standing challenges in regioselective functionalization of pyridine rings, specifically targeting the C4 position while maintaining the integrity of the fluorine and hydroxyl groups. By leveraging a strategic protection-lithiation-deprotection sequence, the disclosed process achieves high purity and yield without the need for transition metal catalysts. For global procurement teams and R&D directors, this represents a pivotal shift towards more sustainable and cost-effective manufacturing of complex fluoropyridine derivatives.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, introducing a carboxylic acid moiety at the C4 position of a 2-fluoro-3-hydroxypyridine scaffold has been fraught with synthetic difficulties. Conventional approaches often rely on multi-step sequences involving harsh oxidation conditions or the use of expensive transition metal catalysts which can leave behind toxic residues requiring extensive purification. Furthermore, the presence of the free hydroxyl group at the C3 position poses a significant compatibility issue with strong organometallic reagents, often leading to decomposition or non-selective side reactions. These inefficiencies result in lower overall yields, increased waste generation, and prolonged production cycles, creating substantial bottlenecks for supply chain managers aiming to secure reliable volumes of high-purity intermediates. The lack of a direct, robust method has historically driven up the cost of goods for downstream API manufacturers.
The Novel Approach
The methodology outlined in patent CN110669002A circumvents these obstacles through an elegant two-step strategy that prioritizes atom economy and operational simplicity. The core innovation lies in the temporary masking of the reactive hydroxyl group using a SEM (2-(trimethylsilyl)ethoxymethyl) protecting group, which renders the molecule compatible with aggressive lithiation conditions. This protection allows for the precise generation of a carbanion at the C4 position using n-butyllithium, followed by immediate quenching with carbon dioxide to install the carboxylic acid functionality. The subsequent acidic workup seamlessly removes the protecting group, delivering the target 2-fluoro-3-hydroxypyridine-4-carboxylic acid in high purity. This streamlined approach not only reduces the number of unit operations but also eliminates the need for chromatographic purification, offering a clear pathway for cost reduction in fine chemical manufacturing.
Mechanistic Insights into SEM-Protection and Directed Lithiation
The success of this synthetic route hinges on the precise control of reactivity through the SEM protection strategy. In the first stage, the phenolic hydroxyl group of 2-fluoro-3-hydroxypyridine acts as a nucleophile, attacking the electrophilic carbon of SEMCl in the presence of DIEA as a base. This transformation converts the acidic proton into a robust silyl ether, which is stable under the strongly basic conditions required for the subsequent step. Without this protection, the hydroxyl proton would be rapidly deprotonated by n-butyllithium, consuming the reagent and preventing the desired C-H activation. The choice of SEM is particularly advantageous due to its stability during lithiation and its ease of removal under mild acidic conditions, ensuring that the final product is obtained without residual protecting group impurities.
Following protection, the mechanism proceeds via a Directed Ortho-Metalation (DoM) pathway facilitated by the pyridine nitrogen atom. Upon treatment with lithium diisopropylamide (LDA), generated in situ from diisopropylamine and n-butyllithium at cryogenic temperatures, the base selectively abstracts the proton at the C4 position. This regioselectivity is driven by the coordination of the lithium cation to the pyridine nitrogen, which directs the base to the adjacent ortho-position relative to the nitrogen, while the C2 position is blocked by the fluorine atom. The resulting C4-lithiated species is then trapped by electrophilic carbon dioxide, forming a carboxylate salt. 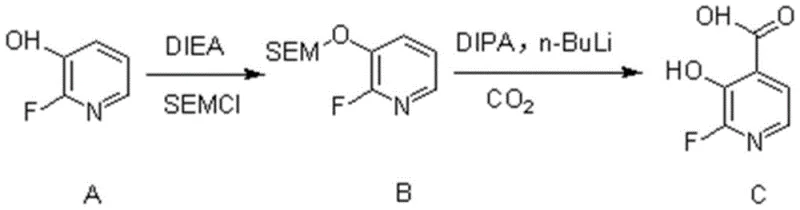 The final acidic workup serves a dual purpose: it protonates the carboxylate to form the free acid and cleaves the SEM ether to regenerate the hydroxyl group, completing the transformation in a highly efficient manner.
The final acidic workup serves a dual purpose: it protonates the carboxylate to form the free acid and cleaves the SEM ether to regenerate the hydroxyl group, completing the transformation in a highly efficient manner.
How to Synthesize 2-Fluoro-3-Hydroxypyridine-4-Carboxylic Acid Efficiently
The practical execution of this synthesis involves dissolving the starting material in dichloromethane and treating it with SEMCl and DIEA at ambient temperature to ensure complete conversion to the protected intermediate. After isolation, the intermediate is subjected to lithiation in tetrahydrofuran at -78°C using freshly prepared LDA, followed by the introduction of carbon dioxide gas to effect carboxylation. The detailed standardized synthesis steps are provided in the guide below.
- Protect the hydroxyl group of 2-fluoro-3-hydroxypyridine using SEMCl and DIEA in dichloromethane to form the SEM-ether intermediate.
- Generate LDA in situ using diisopropylamine and n-butyllithium in THF at -78°C, then add the protected intermediate for regioselective lithiation at the C4 position.
- Quench the lithiated species with carbon dioxide gas, followed by acidic workup to simultaneously remove the protecting group and isolate the final carboxylic acid product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthetic route offers compelling economic and logistical benefits. By simplifying the production process to just two main chemical transformations, manufacturers can significantly reduce the consumption of solvents and reagents, leading to a leaner operational footprint. The elimination of transition metal catalysts removes the costly and time-consuming step of heavy metal scavenging, which is often a regulatory requirement for pharmaceutical intermediates. This streamlining translates directly into improved margin potential and faster time-to-market for clients relying on this key building block.
- Cost Reduction in Manufacturing: The process utilizes inexpensive and widely available reagents such as carbon dioxide and n-butyllithium, avoiding the need for precious metal catalysts like palladium or rhodium which are subject to volatile market pricing. Furthermore, the high yields reported in the patent examples indicate minimal material loss, allowing for better utilization of raw materials and reduced waste disposal costs. The simplicity of the workup procedure, which relies on standard extraction and crystallization techniques rather than complex chromatography, further drives down processing expenses and energy consumption.
- Enhanced Supply Chain Reliability: The starting material, 2-fluoro-3-hydroxypyridine, is a commercially accessible commodity chemical, reducing the risk of supply disruptions associated with custom-synthesized precursors. The robustness of the reaction conditions means that production can be scaled up with high confidence, ensuring consistent availability of the intermediate for downstream customers. By establishing a supply line based on this reliable chemistry, companies can mitigate the risks of delays caused by complex multi-step syntheses or dependency on single-source specialty reagents.
- Scalability and Environmental Compliance: The synthetic route is inherently scalable, as demonstrated by the patent's use of standard laboratory equipment that translates well to industrial reactors. The avoidance of toxic heavy metals aligns with increasingly stringent environmental regulations and green chemistry principles, simplifying the permitting process for manufacturing facilities. Additionally, the use of carbon dioxide as a C1 building block represents a sustainable approach to carbon incorporation, enhancing the environmental profile of the final product and appealing to eco-conscious stakeholders.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of 2-fluoro-3-hydroxypyridine-4-carboxylic acid. These insights are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy and relevance for industry professionals evaluating this technology for their supply chains.
Q: What is the primary advantage of using the SEM protecting group in this synthesis?
A: The SEM (2-(trimethylsilyl)ethoxymethyl) group effectively masks the acidic hydroxyl proton, preventing interference with the strong base n-butyllithium during the subsequent lithiation step, thereby ensuring high regioselectivity at the C4 position.
Q: How does this method improve supply chain reliability for fluoropyridine intermediates?
A: By utilizing readily available starting materials like 2-fluoro-3-hydroxypyridine and common reagents such as CO2 and n-BuLi, the process eliminates dependency on exotic catalysts, significantly reducing lead times and procurement risks.
Q: Is this synthetic route suitable for large-scale commercial production?
A: Yes, the patent describes a concise two-step sequence with mild reaction conditions (25°C for protection, -78°C to ambient for lithiation) and high yields, making it highly amenable to kilogram-to-ton scale manufacturing without complex purification requirements.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Fluoro-3-Hydroxypyridine-4-Carboxylic Acid Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality fluorinated intermediates play in the development of advanced pharmaceuticals and agrochemicals. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements with consistency and precision. We are committed to maintaining stringent purity specifications through our rigorous QC labs, guaranteeing that every batch of 2-fluoro-3-hydroxypyridine-4-carboxylic acid meets the exacting standards required for GMP manufacturing environments.
We invite you to collaborate with us to leverage this innovative synthetic technology for your projects. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume needs. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our optimized manufacturing processes can enhance your supply chain efficiency and reduce your overall cost of goods.