Advanced One-Pot Synthesis of Diaryl-Tetrahydropyridines for Commercial Pharmaceutical Manufacturing
The pharmaceutical and fine chemical industries are constantly seeking more efficient pathways to access complex nitrogen-containing heterocycles, which serve as critical scaffolds for bioactive molecules. Patent CN110407739B introduces a groundbreaking preparation method for (4,6-diaryl-tetrahydropyridin-3-yl)(aryl)methanones that fundamentally shifts the paradigm from multi-step, hazardous syntheses to a streamlined, one-pot catalytic process. This innovation leverages a sophisticated cascade involving Meyer-Schuster rearrangement followed by conjugate additions, driven by Lewis acid catalysis. For R&D directors and procurement specialists, this technology represents a significant leap forward in process chemistry, offering a route that not only simplifies operational complexity but also achieves an impressive atom utilization rate of up to 100%. By eliminating the need for stoichiometric organometallic reagents and reducing waste generation, this method aligns perfectly with modern green chemistry principles while ensuring high purity standards required for reliable pharmaceutical intermediate supplier networks.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the tetrahydropyridine core has been fraught with synthetic challenges that hinder scalability and cost-efficiency. Early methodologies, such as those reported by Piper and Wright, relied heavily on Grignard reactions involving styryl cyanide and phenyl magnesium bromide. As illustrated in the reaction scheme below, these traditional routes necessitate rigorous anhydrous conditions, low temperatures, and the handling of pyrophoric reagents, which pose significant safety risks in a commercial setting.
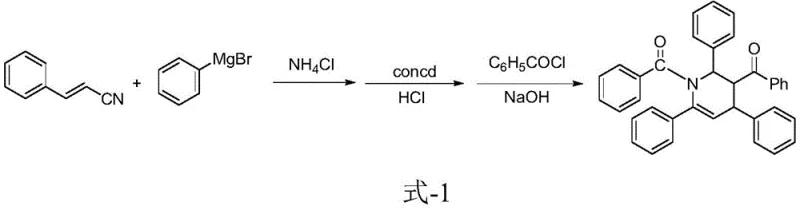
Furthermore, these legacy processes often suffer from poor atom economy due to the generation of stoichiometric salt byproducts and require extensive downstream purification to remove metal residues. Another reported approach by Pavel et al. utilized 1,3,5-triphenyl-2-methylene-1,5-pentanedione as a starting material. However, the synthesis of this specific precursor is itself complex and low-yielding, creating a bottleneck in the supply chain. These conventional methods typically involve multiple isolation steps, leading to cumulative yield losses and increased production costs, making them less attractive for the commercial scale-up of complex pharmaceutical intermediates.
The Novel Approach
In stark contrast, the methodology disclosed in CN110407739B utilizes readily available aryl propiolic alcohols and chalcone derivatives as building blocks. The core innovation lies in the tandem activation of these substrates using a Lewis acid catalyst, facilitating a seamless transformation into the target tetrahydropyridine structure. The general reaction equation demonstrates how three distinct components—alkynol, chalcone, and amine—are integrated in a single vessel without the need for intermediate isolation.

This novel approach operates under mild reflux conditions in common organic solvents, drastically reducing energy consumption and equipment requirements. The reaction proceeds with exceptional efficiency, achieving yields exceeding 73% in optimized examples. By avoiding the use of expensive transition metals or hazardous organometallics, this process significantly lowers the barrier to entry for manufacturing these valuable scaffolds. The simplicity of the workup procedure, which involves basic aqueous quenching and extraction, further enhances its appeal for cost reduction in pharmaceutical intermediate manufacturing, allowing producers to allocate resources more effectively across their portfolio.
Mechanistic Insights into Lewis Acid-Catalyzed Cascade Cyclization
The success of this synthesis hinges on a carefully orchestrated sequence of chemical transformations initiated by the Lewis acid catalyst. The mechanism begins with the activation of the aryl propiolic alcohol, promoting a Meyer-Schuster rearrangement to generate an alpha,beta-unsaturated carbonyl intermediate in situ. This reactive species immediately undergoes a conjugate addition with the chalcone derivative, forming a complex 1,5-dicarbonyl framework. Subsequent intramolecular cyclization with the amine component closes the piperidine ring, finalizing the tetrahydropyridine core. The choice of catalyst, such as bismuth triflate or iron trichloride, is critical as it modulates the electrophilicity of the intermediates without promoting excessive side reactions.

Controlling the impurity profile is paramount for pharmaceutical applications, and this patent provides deep insights into managing side reactions. A key finding is the sensitivity of the reaction to time and catalyst loading. If the reaction time after amine addition is too short, the cyclization remains incomplete, leading to the accumulation of open-chain intermediates. Conversely, prolonged exposure to the acidic environment can trigger degradation or the formation of smaller cyclic byproducts. The patent explicitly identifies a specific non-ring-closed byproduct structure that forms when reaction times are insufficient, highlighting the importance of precise kinetic control.
By optimizing the molar ratio of the acid catalyst to the alkynol substrate, manufacturers can steer the reaction towards either the tetrahydropyridine (Formula I) or the dihydropyridine (Formula II) derivative. This tunability allows for the production of diverse analogues simply by adjusting reaction parameters rather than changing the entire synthetic route. Such mechanistic understanding ensures that the final high-purity pharmaceutical intermediates meet stringent regulatory specifications, minimizing the risk of toxic impurities carrying through to the final drug substance.
How to Synthesize (4,6-diaryl-tetrahydropyridin-3-yl)(aryl)methanone Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires adherence to specific procedural guidelines to maximize yield and reproducibility. The process is designed to be robust, utilizing standard glassware and heating mantles, yet it demands attention to the timing of reagent addition. The initial condensation of the alkynol and chalcone must reach completion before the amine is introduced to prevent competitive side reactions. Detailed standard operating procedures regarding solvent selection, catalyst loading, and thermal profiles are essential for consistent results.
- Mix aryl propiolic alcohol, chalcone derivative, Lewis acid catalyst, and solvent in a sealed tube.
- Heat the mixture under reflux until the alkynol is completely consumed, monitoring via TLC.
- Add the amine compound and continue refluxing for 9 to 17 hours depending on the desired product structure.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this catalytic one-pot method offers tangible strategic benefits beyond mere technical elegance. The primary advantage lies in the drastic simplification of the raw material supply chain. Chalcones and aryl propiolic alcohols are commodity chemicals available from numerous global vendors, reducing dependency on single-source suppliers for exotic precursors. This diversification of the supply base enhances resilience against market fluctuations and logistical disruptions, ensuring reducing lead time for high-purity pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The elimination of stoichiometric Grignard reagents and the associated cryogenic cooling requirements translates directly into lower utility and material costs. Furthermore, the 100% atom economy means that nearly every atom in the starting materials ends up in the final product, virtually eliminating the cost of waste disposal and treatment. The simplified workup procedure reduces solvent consumption and labor hours, contributing to substantial overall cost savings without compromising quality.
- Enhanced Supply Chain Reliability: The mild reaction conditions (reflux in common solvents like dichloroethane or toluene) allow the process to be run in standard stainless steel reactors without the need for specialized Hastelloy lining required for corrosive acids or pyrophoric reagents. This compatibility with existing infrastructure accelerates technology transfer and scale-up timelines. The robustness of the reaction also means fewer batch failures, leading to more predictable delivery schedules for downstream customers.
- Scalability and Environmental Compliance: As regulatory pressure mounts to reduce the environmental footprint of chemical manufacturing, this method stands out for its green credentials. The absence of heavy metal catalysts simplifies the removal of trace metals, a critical step in API synthesis. The high conversion rates minimize the volume of organic waste streams, facilitating easier compliance with environmental regulations. This sustainability profile adds value to the end product, appealing to eco-conscious pharmaceutical partners.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis route. Understanding these nuances helps stakeholders make informed decisions about adopting this technology for their specific production needs. The answers are derived directly from the experimental data and comparative examples provided in the patent documentation.
Q: What are the key advantages of this synthesis method over traditional Grignard routes?
A: Unlike traditional methods requiring harsh Grignard reagents and multiple steps, this patent describes a one-pot process with 100% atom economy, milder conditions, and easier purification.
Q: How is product selectivity controlled between tetrahydropyridine and dihydropyridine derivatives?
A: Selectivity is precisely tuned by adjusting the molar ratio of the acid catalyst and strictly controlling the reaction time after amine addition, ranging from 9 to 17 hours.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the use of commercially available raw materials like chalcones and propargyl alcohols, combined with simple reflux conditions, makes it highly scalable for industrial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable (4,6-diaryl-tetrahydropyridin-3-yl)(aryl)methanone Supplier
The technological advancements detailed in CN110407739B underscore the potential for efficient, high-quality production of complex heterocyclic intermediates. At NINGBO INNO PHARMCHEM, we possess the technical expertise and infrastructure to leverage such innovative synthetic routes for our clients. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that laboratory successes are seamlessly translated into industrial reality. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch meets the highest international standards.
We invite you to explore how this advanced synthesis method can optimize your supply chain and reduce your overall manufacturing costs. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your project requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how we can become your trusted partner in delivering high-value chemical solutions.