Advanced Synthesis and Commercial Supply of Novel Torasemide Impurity TL-Imp07 for Global QC Standards
Advanced Synthesis and Commercial Supply of Novel Torasemide Impurity TL-Imp07 for Global QC Standards
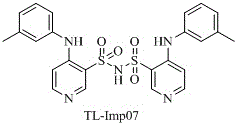
The pharmaceutical industry continuously faces the challenge of identifying and controlling trace impurities in active pharmaceutical ingredients (APIs) to ensure patient safety and regulatory compliance. A significant breakthrough in this domain is documented in patent CN113620868A, which discloses a novel impurity of Torasemide, designated as TL-Imp07, along with a robust preparation method. Torasemide, a highly effective loop diuretic widely used for treating edema and hypertension, requires stringent quality control due to its complex synthesis pathway which can generate various side products. This patent addresses a critical gap by characterizing a previously unreported dimeric impurity, 4-m-toluylamino-N-[(4-m-toluylamino-pyridin-3-yl)sulfonyl]pyridine-3-sulfonamide, providing the industry with a definitive reference standard. The ability to synthesize this specific impurity allows quality control laboratories to accurately quantify its presence in bulk drugs, thereby preventing potential safety issues associated with unknown contaminants. For R&D directors and procurement specialists, understanding the genesis and control of TL-Imp07 is paramount for maintaining supply chain integrity and meeting global pharmacopoeia standards.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the identification of process-related impurities in Torasemide manufacturing has been a reactive and often imprecise endeavor. Conventional quality control relies heavily on detecting known impurities such as EP Impurities A through E, which are well-documented in existing literature. However, the synthesis of Torasemide involves multiple steps including chlorosulfonation, amination, and urea formation, creating ample opportunities for unforeseen side reactions. When unknown peaks appear in HPLC chromatograms during stability testing or batch release, manufacturers often lack authentic reference standards to identify them definitively. This uncertainty forces companies to either reject entire batches conservatively, leading to significant financial loss, or risk releasing products with unidentified genotoxic or toxic impurities. Furthermore, isolating these unknown impurities from crude reaction mixtures for structural elucidation is technically demanding, time-consuming, and yields insufficient quantities for comprehensive toxicological assessment. The absence of a reliable synthetic route for specific dimeric impurities like TL-Imp07 has historically left a blind spot in the impurity profile of this critical diuretic medication.
The Novel Approach
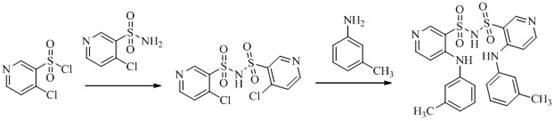
The methodology presented in patent CN113620868A offers a proactive solution by establishing a deliberate synthetic pathway to generate TL-Imp07. Instead of relying on chance isolation from production waste, this approach utilizes a targeted condensation reaction between 4-chloro-3-pyridine sulfonamide and 4-chloro-3-pyridine sulfonyl chloride. This strategic construction of the sulfonamide bridge mimics the potential side reactions occurring during the main API synthesis but under controlled conditions to maximize the formation of the specific dimer. By shifting from passive detection to active synthesis, manufacturers can produce gram-to-kilogram quantities of the impurity standard. This availability empowers analytical teams to spike samples, validate HPLC methods, and set precise acceptance criteria. The novel approach transforms an unknown variable into a controlled parameter, significantly de-risking the regulatory filing process for Torasemide generics and ensuring that the final drug product meets the highest purity specifications required by agencies like the FDA and EMA.
Mechanistic Insights into Sulfonamide Condensation and Ammoniation
The synthesis of TL-Imp07 proceeds through a sophisticated mechanism involving nucleophilic substitution and sulfonylation, key reactions in heterocyclic chemistry. The process initiates with the activation of the sulfonyl group, where 4-chloro-3-pyridine sulfonyl chloride acts as an electrophile. In the presence of a base such as potassium carbonate or triethylamine, the sulfonamide nitrogen of 4-chloro-3-pyridine sulfonamide becomes nucleophilic. This nucleophile attacks the sulfur atom of the sulfonyl chloride, displacing the chloride ion and forming a stable sulfonamide linkage. This step is critical as it creates the central backbone of the TL-Imp07 molecule, effectively dimerizing two pyridine rings through a sulfonyl bridge. The choice of solvent, typically dichloromethane, and the stoichiometry of the base are vital to prevent over-reaction or hydrolysis of the sensitive sulfonyl chloride moiety. Following the formation of the chloro-intermediate, the second stage involves an ammoniation reaction with m-toluidine. Here, the aromatic amine displaces the remaining chlorine atoms on the pyridine rings via nucleophilic aromatic substitution, facilitated by heating in polar aprotic solvents like DMF.
Controlling impurities during this synthesis is achieved through careful modulation of reaction kinetics and purification strategies. The patent highlights the use of column chromatography as a decisive purification step, utilizing a dichloromethane and methanol gradient. This technique effectively separates the target TL-Imp07 from unreacted starting materials, mono-substituted intermediates, and other regioisomers. The high purity achieved (reported up to 97.1% HPLC purity in examples) demonstrates the efficacy of this separation protocol. From a mechanistic standpoint, the stability of the sulfonamide bond ensures that the impurity does not degrade easily during storage, making it an ideal reference standard. Understanding this mechanism allows process chemists to identify critical process parameters (CPPs) in the main Torasemide synthesis that might favor the formation of this dimer, enabling them to tweak reaction conditions to suppress its generation in the commercial API process while simultaneously having the standard ready for QC testing.
How to Synthesize TL-Imp07 Efficiently
The preparation of TL-Imp07 is designed to be operationally simple yet chemically robust, suitable for scaling in a laboratory or pilot plant environment. The process begins with the condensation of equimolar amounts of 4-chloro-3-pyridine sulfonamide and 4-chloro-3-pyridine sulfonyl chloride hydrochloride in dichloromethane. A base, such as potassium carbonate, is added to neutralize the hydrochloride salt and drive the reaction forward at room temperature. After stirring for several hours, the intermediate is isolated via aqueous workup and drying. The second step involves reacting this intermediate with m-toluidine in N,N-dimethylformamide (DMF) at elevated temperatures ranging from 80°C to 90°C. This thermal energy is necessary to overcome the activation barrier for the nucleophilic substitution on the electron-deficient pyridine ring. The detailed standardized synthesis steps, including specific reagent grades and exact workup procedures, are outlined in the guide below to ensure reproducibility and high yield.
- Condense 4-chloro-3-pyridine sulfonamide with 4-chloro-3-pyridine sulfonyl chloride in dichloromethane using a base like potassium carbonate to form the intermediate.
- React the resulting intermediate with m-toluidine in N,N-dimethylformamide (DMF) under heating conditions (80-90°C) to facilitate ammoniation.
- Purify the crude reaction mixture using silica gel column chromatography with a dichloromethane and methanol solvent system to isolate high-purity TL-Imp07.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the availability of a synthesized reference standard for TL-Imp07 translates directly into risk mitigation and cost optimization. The primary advantage lies in the stabilization of the supply chain for Torasemide bulk drugs. By having access to a verified impurity standard, manufacturers can avoid costly batch failures caused by unidentified peaks during quality control testing. This reliability reduces the lead time for batch release, ensuring a continuous flow of product to the market without interruptions due to regulatory queries regarding unknown impurities. Furthermore, the synthesis method described avoids the use of exotic catalysts or extreme conditions, relying on commodity chemicals like m-toluidine and common solvents. This simplicity suggests that the reference standard itself can be sourced at a reasonable cost, avoiding the premium pricing often associated with custom-synthesized rare impurities.
- Cost Reduction in Manufacturing: The implementation of this specific impurity standard allows for more precise quality control, which minimizes the rate of false-positive rejections of API batches. By accurately distinguishing between harmless process variations and genuine out-of-specification impurities, manufacturers can significantly reduce waste and improve overall yield efficiency. Additionally, the synthetic route utilizes inexpensive, readily available starting materials and standard laboratory equipment, avoiding the need for specialized high-pressure reactors or precious metal catalysts. This economic efficiency in producing the standard ensures that the cost of quality control remains low, contributing to the overall cost reduction in pharmaceutical manufacturing operations without compromising on safety or compliance standards.
- Enhanced Supply Chain Reliability: Dependence on isolating impurities from production waste creates a bottleneck, as the availability of the standard is tied to the production schedule of the API itself. The independent synthesis route decouples the supply of the reference standard from the main manufacturing line, ensuring that QC labs always have access to the necessary materials for method validation and routine testing. This independence enhances supply chain resilience, protecting against disruptions in the primary API production that could otherwise halt quality assurance activities. Reliable access to TL-Imp07 ensures that regulatory filings remain robust and that audits can be passed smoothly, securing the long-term continuity of the Torasemide supply for global markets.
- Scalability and Environmental Compliance: The described preparation method is inherently scalable, moving seamlessly from gram-scale laboratory synthesis to kilogram-scale production required for large pharmaceutical companies. The use of standard organic solvents like dichloromethane and DMF allows for established waste management protocols to be applied, ensuring environmental compliance. The purification via column chromatography, while labor-intensive at small scale, can be adapted to flash chromatography or recrystallization techniques at larger scales to improve throughput. This scalability ensures that as the demand for Torasemide grows, the capacity to monitor its quality grows in tandem, supporting sustainable manufacturing practices and adherence to green chemistry principles by minimizing the generation of undefined chemical waste.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the novel Torasemide impurity TL-Imp07. These answers are derived directly from the technical specifications and experimental data provided in patent CN113620868A, offering clarity on its synthesis, application, and regulatory significance. Understanding these details is crucial for stakeholders involved in the quality assurance and procurement of diuretic pharmaceuticals.
Q: What is the chemical structure of the new Torasemide impurity TL-Imp07?
A: TL-Imp07 is chemically defined as 4-m-toluylamino-N-[(4-m-toluylamino-pyridin-3-yl)sulfonyl]pyridine-3-sulfonamide. It represents a dimeric side-product formed during the synthesis of Torasemide involving sulfonamide linkages.
Q: Why is controlling TL-Imp07 critical for Torasemide manufacturing?
A: As a novel process-related impurity, TL-Imp07 can persist in the final bulk drug if not monitored. Identifying and quantifying it ensures compliance with strict ICH guidelines for impurity profiling, guaranteeing patient safety and drug efficacy.
Q: How does the patented synthesis method improve impurity reference standard availability?
A: The patent provides a robust, scalable two-step synthesis route yielding high purity (over 94% HPLC). This allows manufacturers to produce authentic reference standards reliably, replacing uncertain isolation methods from crude reaction mixtures.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable TL-Imp07 Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality impurity standards play in the pharmaceutical lifecycle. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the demands of both early-stage R&D and full-scale commercial manufacturing. We are committed to delivering TL-Imp07 with stringent purity specifications, supported by our rigorous QC labs that utilize state-of-the-art analytical instrumentation. Our capability to replicate the patented synthesis ensures a consistent supply of this reference standard, empowering your quality control teams to maintain the highest levels of drug safety and efficacy.
We invite you to optimize your supply chain by partnering with us for your specialized chemical needs. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our TL-Imp07 supply can enhance your operational efficiency and regulatory compliance.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →