Advanced Synthetic Route for [1,5-a]-Pyridoimidazole-1-Carbonitrile: Technical Insights for Commercial Scale-Up
Introduction to Novel Heterocyclic Synthesis Technologies
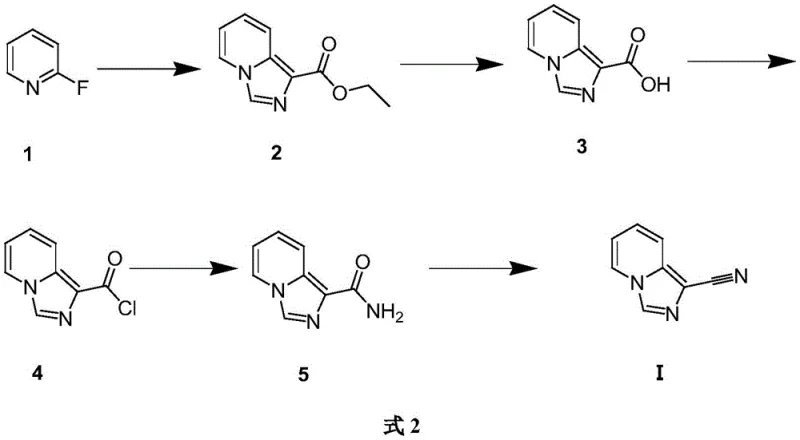
The pharmaceutical and fine chemical industries are constantly seeking robust, scalable pathways for complex heterocyclic intermediates that serve as critical building blocks for next-generation therapeutics. Patent CN110590771B introduces a significant advancement in this domain by detailing a highly efficient chemical synthesis method for [1,5-a]-pyridoimidazole-1-carbonitrile. This compound, characterized by its fused pyridine-imidazole core structure, represents a valuable scaffold in medicinal chemistry, potentially applicable in the development of antiviral, antibacterial, and antitumor agents. The disclosed methodology leverages 2-fluoropyridine as a commercially accessible starting material, transforming it through a logical sequence of five distinct chemical transformations. By optimizing reaction conditions such as temperature control during the critical cyclization and dehydration steps, this patent provides a reproducible framework that addresses common challenges in heterocyclic synthesis, including yield optimization and impurity management.
![Chemical structure of [1,5-a]-pyridoimidazole-1-carbonitrile showing the fused ring system and nitrile group](/insights/img/pyridoimidazole-carbonitrile-synthesis-pharma-supplier-20260303023638-01.png)
For R&D directors and process chemists, the structural integrity and functional group tolerance of this molecule are paramount. The presence of the nitrile group at the 1-position of the imidazo[1,5-a]pyridine ring system offers a versatile handle for further derivatization, making it an ideal candidate for library synthesis in drug discovery programs. The patent emphasizes a stepwise approach that balances reactivity with selectivity, ensuring that the sensitive nitrogen-containing rings are constructed without excessive degradation. This technical foundation sets the stage for a deeper analysis of how this specific route compares to conventional methods and why it holds substantial promise for commercial adoption by reliable pharmaceutical intermediate suppliers.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the synthesis of fused imidazo-pyridine derivatives has often been plagued by inefficient routes that rely on scarce starting materials or require extreme reaction conditions that are difficult to maintain on a large scale. Many legacy processes involve multi-step sequences with poor atom economy, where the introduction of the imidazole ring necessitates harsh cyclization agents or precious metal catalysts that drive up costs and complicate downstream purification. Furthermore, conventional methods frequently struggle with regioselectivity issues, leading to complex mixtures of isomers that are arduous to separate, thereby reducing the overall yield of the desired pharmacophore. The reliance on unstable intermediates in older protocols also poses significant safety risks and supply chain vulnerabilities, as these materials often have short shelf lives and require specialized storage conditions. Consequently, procurement managers have historically faced challenges in securing consistent, high-quality supplies of such intermediates due to the inherent instability and variability of these traditional manufacturing processes.
The Novel Approach
In stark contrast, the methodology outlined in CN110590771B presents a streamlined, five-step trajectory that circumvents many of these historical bottlenecks by utilizing 2-fluoropyridine, a stable and widely available commodity chemical. This novel approach capitalizes on the nucleophilic displacement of the fluorine atom to initiate ring closure, a strategy that is both mechanistically sound and operationally simple. By breaking the synthesis down into discrete stages—cyclization, hydrolysis, acyl halide formation, amidation, and dehydration—the process allows for rigorous quality control at each juncture, ensuring that impurities do not carry over into the final product. The use of standard reagents such as sodium hydride, thionyl chloride, and phosphorus oxychloride means that the process can be easily implemented in existing multipurpose chemical plants without the need for exotic equipment. This shift towards a more modular and robust synthetic design significantly enhances the feasibility of cost reduction in API manufacturing, as it minimizes waste generation and maximizes the throughput of the final nitrile product.
Mechanistic Insights into the Five-Step Cascade Synthesis
The core of this technological breakthrough lies in the precise orchestration of reaction mechanisms that build the fused ring system with high fidelity. The initial step involves the deprotonation of ethyl cyanoacetate by sodium hydride in dimethyl sulfoxide (DMSO), generating a reactive nucleophile that attacks the electron-deficient carbon adjacent to the fluorine in 2-fluoropyridine. This nucleophilic aromatic substitution is followed by an intramolecular cyclization to form the imidazo[1,5-a]pyridine core, a critical transformation that establishes the scaffold's geometry. Subsequent hydrolysis of the ethyl ester moiety under basic conditions, followed by acidification, cleanly yields the corresponding carboxylic acid, which serves as the pivot point for functional group manipulation. The conversion of this acid to an acid chloride using thionyl chloride activates the carbonyl carbon, facilitating the subsequent nucleophilic attack by ammonia to form the primary amide. Finally, the dehydration of this amide using phosphorus oxychloride eliminates water to install the crucial nitrile functionality, completing the synthesis of the target molecule.
From an impurity control perspective, the mechanism offers several advantages that are critical for producing high-purity pharmaceutical intermediates. The intermediate isolation steps, particularly the precipitation of the carboxylic acid and the filtration of the amide, act as effective purification gates that remove side products and unreacted starting materials before they can interfere with subsequent reactions. For instance, the hydrolysis step is conducted at controlled temperatures ranging from 30°C to 100°C, which prevents the degradation of the sensitive heterocyclic ring while ensuring complete conversion of the ester. Similarly, the final dehydration step is carefully managed between 50°C and 110°C to drive the elimination reaction to completion without causing polymerization or decomposition of the nitrile product. This meticulous attention to thermal parameters and reaction stoichiometry ensures that the final product meets stringent purity specifications required for downstream drug synthesis, thereby reducing the burden on analytical QC labs.
How to Synthesize [1,5-a]-Pyridoimidazole-1-Carbonitrile Efficiently
Implementing this synthesis route requires a disciplined approach to process parameters, particularly regarding temperature control and reagent addition rates to manage exotherms and ensure safety. The patent details a specific protocol where the initial cyclization is performed under nitrogen protection to prevent moisture interference, followed by a quenching and extraction sequence that isolates the ethyl ester intermediate. The subsequent transformation to the acid chloride and amide requires careful handling of corrosive reagents like thionyl chloride and ammonia water, emphasizing the need for appropriate corrosion-resistant reactor materials. Detailed standardized synthesis steps see the guide below for operational specifics.
- Cyclization of 2-fluoropyridine with ethyl cyanoacetate using sodium hydride in DMSO to form the ethyl ester intermediate.
- Hydrolysis of the ethyl ester using an inorganic base followed by acidification to yield the carboxylic acid.
- Conversion of the carboxylic acid to acid chloride using thionyl chloride, followed by amidation with ammonia water.
- Final dehydration of the amide using phosphorus oxychloride to obtain the target nitrile compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthesis route offers tangible strategic benefits that extend beyond mere technical feasibility. The reliance on 2-fluoropyridine as a feedstock is a major advantage, as it is a bulk chemical produced by numerous global manufacturers, ensuring a stable and competitive supply base that mitigates the risk of raw material shortages. Unlike processes that depend on proprietary or custom-synthesized precursors, this method leverages a commodity supply chain, which inherently drives down costs and improves negotiation leverage for purchasing departments. Furthermore, the simplicity of the workup procedures, which primarily involve standard liquid-liquid extractions and filtrations, reduces the demand for specialized separation technologies, thereby lowering capital expenditure requirements for manufacturing partners.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the use of cost-effective reagents like sodium hydride and phosphorus oxychloride significantly lower the direct material costs associated with production. By avoiding complex catalytic cycles that require ligand recovery or metal scavenging, the process simplifies the bill of materials and reduces waste disposal costs related to heavy metal contamination. Additionally, the high yields reported in the later stages of the synthesis, particularly the hydrolysis and dehydration steps, contribute to a more favorable overall mass balance, ensuring that a greater proportion of input materials are converted into saleable product. This efficiency translates directly into improved margin potential for contract manufacturing organizations and ultimate cost savings for the end-user.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions, which tolerate a range of temperatures and utilize common solvents like DMSO and dichloromethane, ensures that the process can be reliably transferred between different manufacturing sites without significant re-optimization. This flexibility is crucial for maintaining supply continuity in the face of regional disruptions or capacity constraints at specific facilities. The ability to source reagents from multiple vendors further de-risks the supply chain, preventing single points of failure that could lead to production delays. Consequently, this synthesis method supports a resilient supply network capable of meeting the fluctuating demands of the pharmaceutical market.
- Scalability and Environmental Compliance: The stepwise nature of the synthesis allows for easy scale-up from laboratory benchtop quantities to multi-ton commercial production, as each unit operation is well-understood and manageable in large reactors. The process generates waste streams that are largely composed of inorganic salts and common organic solvents, which can be treated using standard effluent treatment protocols, facilitating compliance with increasingly stringent environmental regulations. By minimizing the generation of hazardous byproducts and avoiding the use of persistent organic pollutants, this method aligns with green chemistry principles, enhancing the sustainability profile of the supply chain and appealing to environmentally conscious stakeholders.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of [1,5-a]-pyridoimidazole-1-carbonitrile, based on the detailed disclosures within the patent literature. Understanding these aspects is essential for stakeholders evaluating the integration of this intermediate into their broader development pipelines. The answers provided reflect the specific process parameters and advantages highlighted in the intellectual property documentation.
Q: What is the primary starting material for synthesizing [1,5-a]-pyridoimidazole-1-carbonitrile?
A: The synthesis utilizes 2-fluoropyridine as the primary raw material, which undergoes a nucleophilic substitution and cyclization reaction with ethyl cyanoacetate.
Q: How does this synthesis method improve impurity control?
A: The method employs distinct purification steps such as aqueous workups, extractions, and recrystallization (pulping) between stages, ensuring high purity of the final nitrile product by removing unreacted intermediates and byproducts.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process uses common industrial reagents like thionyl chloride and phosphorus oxychloride and relies on standard unit operations like reflux and filtration, making it highly adaptable for commercial scale-up from kilograms to metric tons.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable [1,5-a]-Pyridoimidazole-1-Carbonitrile Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in accelerating drug discovery and development timelines. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements with consistency and precision. We are committed to delivering [1,5-a]-pyridoimidazole-1-carbonitrile with stringent purity specifications, supported by our rigorous QC labs that employ advanced analytical techniques to verify identity and assay. Our facility is equipped to handle the specific reagents and conditions required for this synthesis, guaranteeing a secure and compliant manufacturing environment.
We invite you to collaborate with us to optimize your supply chain for this valuable heterocyclic building block. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific project needs. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can support your long-term strategic goals in pharmaceutical innovation.