Advanced Synthesis of Palovir Intermediate via Protected Bromoethylamine Alkylation
The pharmaceutical industry continuously seeks robust synthetic routes for complex antiviral agents, and the preparation of palovir intermediates represents a critical challenge in modern medicinal chemistry. Patent CN114605301A discloses a groundbreaking methodology for synthesizing methyl (S)-2-(Boc-amino)-3-[(S)-2-oxo-3-pyrrolidinyl]propionate, a key building block for next-generation therapeutics. This innovative approach fundamentally reimagines the construction of the pyrrolidinyl core by replacing traditional, hazardous nitrile reduction pathways with a highly selective alkylation strategy using protected bromoethylamine derivatives. By leveraging the steric properties of Cbz or Fmoc protecting groups, the process achieves exceptional diastereoselectivity under mild thermal conditions, effectively bypassing the need for extreme cryogenic control often seen in legacy methods. For R&D directors and process chemists, this represents a significant leap forward in impurity control and operational safety, offering a streamlined path from commercially available glutamic acid derivatives to high-value chiral intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of this specific pyrrolidinyl scaffold has relied heavily on routes involving bromoacetonitrile, a highly irritant and toxic reagent that poses significant handling risks in large-scale manufacturing environments. As illustrated in the prior art reaction schemes, these conventional pathways typically necessitate a subsequent reduction of the nitrile group to an amine, a step that is fraught with chemical and environmental challenges. 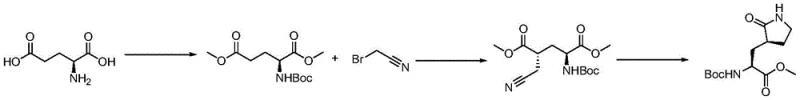 Traditional hydrogenation methods for this reduction require noble metal catalysts like PtO2 or Pd/C under elevated pressures of 3-5 atmospheres, demanding specialized high-pressure equipment and rigorous safety protocols. Alternatively, chemical reduction using sodium borohydride and cobalt chloride generates substantial volumes of wastewater contaminated with heavy metal cobalt ions, creating a severe burden for environmental compliance and waste treatment facilities. Furthermore, the selectivity in these older routes is heavily dependent on maintaining strictly low temperatures around -78°C to minimize diastereomeric impurities, which drastically increases energy consumption and limits reactor throughput.
Traditional hydrogenation methods for this reduction require noble metal catalysts like PtO2 or Pd/C under elevated pressures of 3-5 atmospheres, demanding specialized high-pressure equipment and rigorous safety protocols. Alternatively, chemical reduction using sodium borohydride and cobalt chloride generates substantial volumes of wastewater contaminated with heavy metal cobalt ions, creating a severe burden for environmental compliance and waste treatment facilities. Furthermore, the selectivity in these older routes is heavily dependent on maintaining strictly low temperatures around -78°C to minimize diastereomeric impurities, which drastically increases energy consumption and limits reactor throughput.
The Novel Approach
In stark contrast, the novel methodology disclosed in the patent utilizes N-protected bromoethylamine (either Cbz or Fmoc protected) to directly alkylate the glutamate backbone, fundamentally altering the reaction landscape. 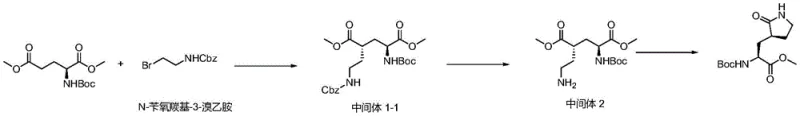 This strategic substitution introduces significant steric bulk near the reactive center, which kinetically favors attack from the less hindered face of the molecule, thereby enhancing the selectivity for the desired (2S, 4S) stereoisomer without the need for extreme cryogenic conditions. The reaction can proceed efficiently at temperatures ranging from -50°C to 0°C, significantly relaxing the thermal constraints and allowing for more flexible process control. Moreover, the removal of the protecting group is achieved under exceptionally mild conditions; for the Cbz variant, simple catalytic hydrogenation at one atmosphere suffices, while the Fmoc variant utilizes standard piperidine treatment. This elimination of high-pressure reduction steps and toxic nitrile reagents results in a cleaner, safer, and more environmentally sustainable process that is ideally suited for commercial scale-up.
This strategic substitution introduces significant steric bulk near the reactive center, which kinetically favors attack from the less hindered face of the molecule, thereby enhancing the selectivity for the desired (2S, 4S) stereoisomer without the need for extreme cryogenic conditions. The reaction can proceed efficiently at temperatures ranging from -50°C to 0°C, significantly relaxing the thermal constraints and allowing for more flexible process control. Moreover, the removal of the protecting group is achieved under exceptionally mild conditions; for the Cbz variant, simple catalytic hydrogenation at one atmosphere suffices, while the Fmoc variant utilizes standard piperidine treatment. This elimination of high-pressure reduction steps and toxic nitrile reagents results in a cleaner, safer, and more environmentally sustainable process that is ideally suited for commercial scale-up.
Mechanistic Insights into LiHMDS-Mediated Alkylation and Cyclization
The core of this synthetic breakthrough lies in the precise control of enolate chemistry using lithium hexamethyldisilazide (LiHMDS) as a strong, non-nucleophilic base. In the initial step, LiHMDS quantitatively deprotonates the alpha-position of the N-Boc-glutamic acid dimethyl ester in tetrahydrofuran, generating a stable lithium enolate species. The subsequent addition of the protected bromoethylamine electrophile triggers an SN2 substitution reaction where the steric bulk of the Cbz or Fmoc group plays a pivotal role in directing the stereochemical outcome. Unlike smaller electrophiles that might allow for non-selective attack, the bulky protecting group creates a defined steric environment that forces the incoming nucleophile to approach from the specific trajectory required to form the (2S, 4S) configuration. This inherent stereocontrol mechanism ensures that the crude product is enriched with the target diastereomer, often eliminating the need for difficult chromatographic purification at this stage and preserving overall yield.
Following the alkylation, the transformation proceeds through a deprotection-cyclization sequence that is both elegant and efficient. In the Cbz pathway, the benzyloxycarbonyl group is cleaved via hydrogenolysis using a catalytic amount of Pd/C under a single atmosphere of hydrogen, a process that is remarkably clean and allows for the recovery and reuse of the precious metal catalyst. The resulting free amine intermediate then undergoes spontaneous or base-promoted intramolecular cyclization. When triethylamine or piperidine is introduced, the free amine attacks the proximal methyl ester carbonyl, closing the five-membered pyrrolidinone ring with the expulsion of methanol. This cascade effectively constructs the complex heterocyclic core in a single pot or telescoped sequence, minimizing unit operations and solvent usage while maintaining high chemical fidelity and purity profiles essential for pharmaceutical applications.
How to Synthesize Palovir Intermediate Efficiently
The synthesis of this high-value intermediate is designed for operational simplicity and robustness, making it accessible for both laboratory optimization and industrial production. The process begins with the preparation of the lithium enolate followed by the controlled addition of the protected electrophile, ensuring that the exothermic nature of the reaction is managed effectively. Detailed standardized operating procedures regarding stoichiometry, addition rates, and quenching protocols are critical for maximizing the diastereomeric ratio and overall yield. The subsequent deprotection and cyclization steps are equally straightforward, utilizing common reagents and standard workup techniques that do not require exotic equipment. For a comprehensive, step-by-step guide including specific molar ratios, solvent volumes, and isolation techniques, please refer to the structured protocol below.
- React N-tert-butyloxycarbonyl-L-glutamic acid dimethyl ester with protected bromoethylamine (Cbz or Fmoc) using LiHMDS in THF at -50°C to 0°C to form the alkylated intermediate.
- Remove the amine protecting group from the intermediate using catalytic hydrogenation (for Cbz) or piperidine treatment (for Fmoc) to generate the free amine precursor.
- Perform intramolecular cyclization by treating the free amine intermediate with a base such as triethylamine or piperidine under reflux or room temperature conditions.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this novel synthetic route offers transformative advantages that directly impact the bottom line and operational reliability. By shifting away from hazardous reagents like bromoacetonitrile and osmium tetroxide (used in alternative oxidative routes), the process significantly reduces the regulatory burden and safety costs associated with handling toxic materials. The elimination of high-pressure hydrogenation steps removes the need for specialized autoclaves, allowing the reaction to be performed in standard glass-lined or stainless steel reactors, which enhances equipment utilization rates and flexibility. Furthermore, the ability to recycle the palladium catalyst in the Cbz deprotection step provides a tangible reduction in raw material costs, as precious metals represent a significant expense in fine chemical manufacturing. These factors combine to create a supply chain that is not only more cost-effective but also more resilient to disruptions caused by regulatory changes or raw material shortages.
- Cost Reduction in Manufacturing: The process achieves substantial cost savings by eliminating the need for expensive noble metal catalysts in high quantities and avoiding the complex waste treatment systems required for heavy metal contaminants like cobalt. The mild reaction conditions reduce energy consumption for cooling and heating, while the high selectivity minimizes the loss of valuable starting materials to side products. Additionally, the potential to skip purification of the intermediate 1 crude product due to high selectivity further streamlines the workflow, reducing solvent usage and processing time, which translates directly into lower manufacturing costs per kilogram of the final API intermediate.
- Enhanced Supply Chain Reliability: The reliance on stable, commercially available starting materials such as N-Boc-glutamic acid dimethyl ester and protected bromoethylamines ensures a secure supply base that is less susceptible to market volatility. The simplified process flow, with fewer unit operations and no requirement for extreme cryogenic infrastructure, allows for faster batch turnover and increased production capacity. This operational agility enables suppliers to respond more rapidly to fluctuating demand from downstream pharmaceutical clients, ensuring consistent delivery schedules and reducing the risk of stockouts that could delay critical drug development programs.
- Scalability and Environmental Compliance: The synthetic route is inherently scalable, having been demonstrated to work efficiently without the constraints of high-pressure equipment or toxic oxidants. The reduction in hazardous waste generation, particularly the avoidance of heavy metal-laden wastewater, simplifies environmental compliance and lowers disposal costs. This green chemistry profile aligns perfectly with the increasing global emphasis on sustainable manufacturing practices, making the facility more attractive to eco-conscious partners and ensuring long-term viability in a tightening regulatory landscape.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis method. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on process parameters and expected outcomes. Understanding these details is crucial for technical teams evaluating the feasibility of adopting this route for their specific production needs.
Q: Why is protected bromoethylamine preferred over bromoacetonitrile in this synthesis?
A: Protected bromoethylamine offers superior steric hindrance, leading to higher diastereoselectivity for the target (2S, 4S) configuration without requiring extreme cryogenic temperatures like -78°C. Additionally, it eliminates the need for hazardous cyano reduction steps involving heavy metals or high-pressure hydrogenation.
Q: Can the palladium catalyst be recycled in the Cbz deprotection step?
A: Yes, the process utilizes a catalytic amount of Pd/C under one atmosphere of hydrogen for debenzylation. The patent explicitly states that the catalyst can be filtered and recycled for reuse, significantly reducing heavy metal waste and operational costs.
Q: What are the advantages of the Fmoc protection strategy compared to Cbz?
A: The Fmoc route allows for deprotection using piperidine in DMF, which can subsequently serve as the base for the final cyclization step. This telescoping capability simplifies the workflow by combining deprotection and ring-closing into a streamlined sequence.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Palovir Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-quality intermediates in the development of life-saving antiviral therapies. Our team of expert process chemists has extensively analyzed this patented route and possesses the technical capability to implement it with precision and efficiency. We boast extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with consistency and reliability. Our state-of-the-art facilities are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of palovir intermediate meets the highest standards required for clinical and commercial applications.
We invite you to collaborate with us to leverage this advanced synthetic technology for your projects. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this optimized route can reduce your overall cost of goods. Please contact us today to request specific COA data and route feasibility assessments, and let us partner with you to accelerate your drug development timeline with a secure and efficient supply chain solution.