Advanced Maravirol Synthesis Technology: Enabling Commercial-Scale Production of High-Purity HIV Treatment API
The patent CN113549070B introduces an innovative synthetic methodology for maravirol and its derivatives, representing a significant advancement in the production of CCR5 receptor antagonists used in HIV treatment regimens. This novel approach utilizes γ-halogen azide compounds as starting materials, enabling a streamlined synthesis pathway that overcomes critical limitations of conventional manufacturing processes. The patented technology delivers substantial improvements in operational efficiency while maintaining stringent pharmaceutical quality requirements essential for antiviral therapeutics. By eliminating multiple protection/deprotection steps required in traditional methods, this process achieves higher overall yields through fewer synthetic transformations while ensuring consistent product purity profiles required for clinical applications. The methodology represents a strategic advancement for pharmaceutical manufacturers seeking reliable production capabilities for critical HIV treatment components.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthesis routes for maravirol typically begin with 3-amino-3-phenylpropionic acid methyl ester, requiring multiple protection/deprotection steps that significantly increase process complexity and reduce overall efficiency. These conventional approaches necessitate careful amino group protection before ester reduction to aldehyde functionality, followed by reductive coupling with complex heterocyclic components and subsequent deprotection steps before final amide formation with 4,4-difluorocyclohexylcarboxylic acid. Each protection/deprotection cycle introduces additional processing time, increases solvent consumption, generates more waste streams, and creates opportunities for impurity formation that complicate purification requirements. The multi-step nature of these traditional routes also leads to cumulative yield losses that substantially impact commercial viability at scale, while the harsh conditions required for certain transformations can compromise product stability and purity profiles essential for pharmaceutical applications.
The Novel Approach
The patented methodology described in CN113549070B fundamentally reimagines the synthetic pathway by starting with γ-halogen azide compounds that enable direct conversion through reduction-hydrolysis-substitution sequences without intermediate purification needs. This innovative approach eliminates multiple protection/deprotection steps required in conventional methods by strategically designing the synthetic sequence around the inherent reactivity of azide functionality. The process can proceed through four distinct but equally viable pathways (Methods A-D), each offering flexibility based on available starting materials and manufacturing constraints while maintaining consistent quality output. By utilizing mild reaction conditions throughout the sequence—typically between 0°C and room temperature—the method achieves superior control over side reactions while significantly reducing energy consumption compared to traditional high-temperature processes. This strategic re-engineering of the synthetic route delivers substantial operational advantages including reduced processing time, lower solvent consumption, simplified purification requirements, and enhanced overall yield profiles.
Mechanistic Insights into Azide-Based Maravirol Synthesis
The core innovation lies in the strategic utilization of γ-halogen azide compounds as versatile starting materials that enable multiple synthetic pathways through controlled manipulation of functional group reactivity. The azide functionality serves as a masked amine that can be selectively reduced under mild conditions using common reducing agents like LiAlH4, NaBH4, or catalytic hydrogenation without affecting other sensitive functional groups present in the molecule. This selective reduction generates primary amine intermediates that readily participate in condensation reactions with carboxylic acid derivatives under standard coupling conditions using triethylamine as base. The halogen substituent at the γ-position provides additional synthetic flexibility by enabling nucleophilic substitution pathways when needed, creating multiple entry points into the synthetic sequence depending on available starting materials and manufacturing constraints.
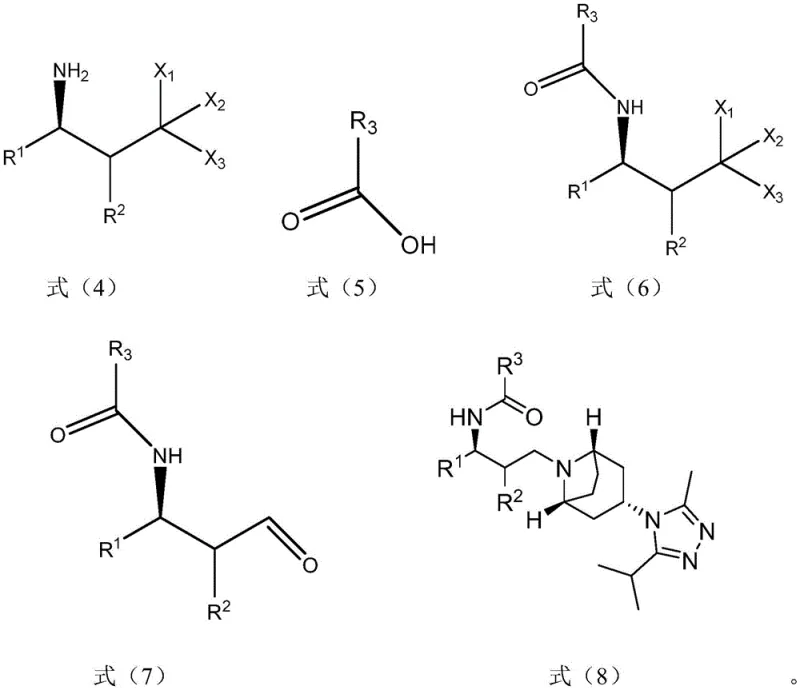
The mechanism demonstrates remarkable versatility through four distinct synthetic routes (Methods A-D), each strategically designed to optimize different aspects of the manufacturing process while maintaining consistent product quality. Method A begins with reduction of the azide compound followed by condensation with carboxylic acid derivatives, hydrolysis to generate aldehyde functionality, and final reductive amination with the complex heterocyclic component. Method B starts with hydrolysis of the azide compound directly to aldehyde functionality before proceeding through reductive amination and subsequent reduction/condensation steps. Each pathway has been carefully engineered to minimize side reactions while maximizing yield and purity through controlled manipulation of functional group reactivity at each stage.
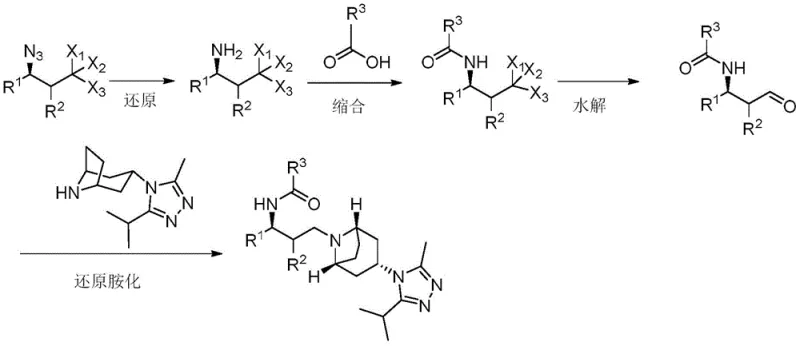
Impurity control is achieved through precise management of reaction conditions at each stage—particularly during hydrolysis steps where silver nitrate catalysis enables selective transformation at mild temperatures (0-70°C) without significant decomposition or side product formation. The elimination of harsh acidic or basic conditions required in conventional methods prevents common degradation pathways that typically generate difficult-to-remove impurities in pharmaceutical manufacturing. This controlled approach ensures consistent product quality while simplifying downstream purification requirements—typically requiring only standard column chromatography rather than more complex purification techniques needed when multiple impurities are present.
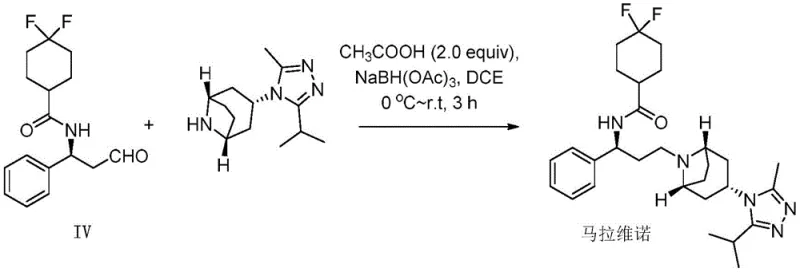
How to Synthesize Maravirol Efficiently
This patented methodology provides pharmaceutical manufacturers with flexible synthetic pathways that can be optimized based on available resources and production requirements while maintaining consistent quality output essential for HIV treatment APIs. The process begins with selection of appropriate starting materials based on commercial availability and cost considerations, followed by implementation of one of four validated synthetic routes that have been specifically designed to maximize efficiency while minimizing impurity formation. Each pathway has been thoroughly validated through experimental work demonstrating consistent yields between 60-87% with purity levels exceeding 98%—exceeding typical industry requirements for pharmaceutical intermediates. Detailed standardized synthesis procedures are available for implementation across various production scales from laboratory development through commercial manufacturing.
- Perform reduction of γ-halogen azide compound using LiAlH4 or NaBH4 under controlled temperature conditions to generate amine intermediate
- Conduct condensation reaction between amine intermediate and carboxylic acid derivative using appropriate coupling agents
- Execute hydrolysis followed by reductive amination with (1R,3s,5S)-3-(3-isopropyl-5-methyl-4H-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]octane
Commercial Advantages for Procurement and Supply Chain Teams
This innovative synthesis methodology delivers substantial commercial benefits that directly address critical pain points experienced by procurement and supply chain teams managing API production for HIV treatment regimens. By streamlining the synthetic sequence and eliminating multiple processing steps required in conventional approaches, this method significantly reduces overall manufacturing complexity while improving resource utilization across all production stages. The elimination of protection/deprotection cycles not only reduces processing time but also minimizes solvent consumption and waste generation—factors that directly impact both operational costs and environmental compliance metrics increasingly important in modern pharmaceutical manufacturing.
- Cost Reduction in Manufacturing: The elimination of multiple protection/deprotection steps reduces overall processing time while minimizing solvent consumption and waste generation across all production stages. By utilizing readily available starting materials and common reagents like triethylamine instead of expensive catalysts or specialized equipment, this methodology achieves significant cost optimization without compromising product quality or regulatory compliance requirements.
- Enhanced Supply Chain Reliability: The flexibility of having four validated synthetic pathways provides manufacturers with strategic options when facing raw material availability challenges or supply chain disruptions. This multi-pathway approach ensures continuous production capability even when specific starting materials experience temporary shortages—significantly improving supply continuity compared to single-pathway conventional methods that lack such strategic flexibility.
- Scalability and Environmental Compliance: The mild reaction conditions (typically between 0°C and room temperature) enable straightforward scale-up from laboratory development through commercial production without requiring specialized high-pressure or high-temperature equipment typically needed for conventional methods. The reduced solvent consumption and waste generation inherent in this streamlined process also align with increasingly stringent environmental regulations while supporting corporate sustainability initiatives.
Frequently Asked Questions (FAQ)
The following questions address common concerns raised by technical procurement teams evaluating this patented methodology for potential implementation in their manufacturing operations. These answers are based on detailed analysis of experimental data presented in patent CN113549070B along with practical considerations from pharmaceutical manufacturing experience.
Q: How does this patented method improve upon conventional maravirol synthesis approaches?
A: The patented method eliminates multiple protection/deprotection steps required in conventional approaches by utilizing γ-halogen azide compounds as starting materials, enabling direct conversion through reduction-hydrolysis-substitution sequences without intermediate purification needs.
Q: What are the key advantages of this synthesis route for commercial manufacturing?
A: This method offers significantly reduced processing time through streamlined reaction sequences, eliminates expensive transition metal catalysts requiring complex removal procedures, and achieves higher purity profiles through fewer side reactions during synthesis.
Q: How does this process address supply chain challenges for HIV treatment APIs?
A: The methodology utilizes readily available starting materials with simplified purification requirements, enabling more reliable supply continuity while maintaining stringent purity specifications required for antiviral pharmaceuticals.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Maravirol Supplier
NINGBO INNO PHARMCHEM brings extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production of complex pharmaceutical intermediates like maravirol while maintaining stringent purity specifications required for HIV treatment APIs. Our state-of-the-art manufacturing facilities feature rigorous QC labs equipped with advanced analytical instrumentation capable of detecting impurities at trace levels—ensuring consistent product quality that meets or exceeds global regulatory requirements across all production scales. As your CDMO partner, we provide comprehensive technical support throughout process development and scale-up phases while implementing robust quality management systems that guarantee reliable supply continuity for critical antiviral therapeutics.
We invite you to request our Customized Cost-Saving Analysis tailored specifically to your manufacturing requirements by contacting our technical procurement team directly—they will provide detailed COA data and route feasibility assessments demonstrating how our patented methodology can enhance your supply chain reliability while optimizing production costs for maravirol-based HIV treatments.