Revolutionizing Fluoroalkyl-Substituted Furan Production: Scalable, Metal-Free Synthesis for Pharmaceutical Intermediates
The patent CN110357842B introduces a groundbreaking methodology for synthesizing fluoroalkyl-substituted furan compounds through a selective cleavage of four carbon-fluorine bonds. This innovative approach represents a significant advancement in heterocyclic chemistry by enabling the direct incorporation of fluorinated moieties into furan scaffolds without requiring harsh reaction conditions or toxic catalysts. The process utilizes readily available polyfluorinated peroxygen compounds and organic sulfinates under mild conditions, offering substantial improvements over conventional furan synthesis techniques that often rely on strong acids, bases, or transition metal catalysts. This novel methodology addresses critical limitations in current synthetic approaches while providing exceptional functional group tolerance and substrate universality essential for complex pharmaceutical intermediate production. The patent demonstrates how this technique can produce high-purity compounds with yields exceeding 99% under optimized conditions, making it particularly valuable for applications requiring stringent purity specifications in pharmaceutical manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional furan synthesis methods such as Feist-Benary, Paal-Knorr, and Diels-Alder approaches suffer from significant drawbacks that limit their practical utility in modern pharmaceutical manufacturing environments. These conventional techniques typically require harsh reaction conditions involving strong acids or bases that complicate process safety and waste management protocols. Furthermore, they exhibit poor functional group compatibility and limited substrate scope, often generating numerous by-products that necessitate extensive purification procedures and reduce overall process efficiency. The reliance on transition metal catalysts in many contemporary methods introduces additional challenges related to metal contamination in final products, which is particularly problematic for pharmaceutical applications requiring ultra-high purity standards. These limitations collectively result in higher production costs, longer development timelines, and compromised supply chain reliability when manufacturing complex furan-based intermediates for drug development pipelines.
The Novel Approach
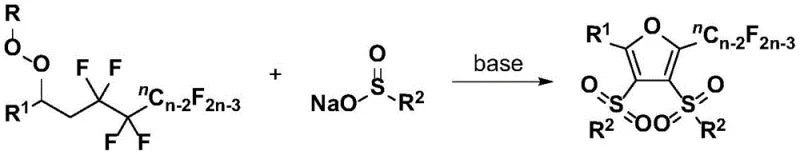
The patented methodology overcomes these longstanding challenges through an elegant three-component coupling system that operates under remarkably mild conditions without transition metal catalysts. By utilizing polyfluorinated peroxygen compounds and organic sulfinates with simple inorganic base promoters like cesium carbonate in tert-butanol solvent at just 50°C, this process achieves exceptional selectivity while maintaining broad functional group tolerance. The reaction proceeds efficiently in air atmosphere without requiring inert gas protection or specialized equipment, significantly simplifying scale-up procedures and reducing capital investment requirements. This innovative approach demonstrates remarkable substrate universality across diverse aromatic systems while producing minimal by-products, thereby streamlining purification workflows and enhancing overall process efficiency. The elimination of toxic metal catalysts not only improves product purity but also addresses critical environmental compliance concerns associated with traditional synthetic routes.
Mechanistic Insights into Metal-Free Radical Coupling
The core innovation lies in a sophisticated radical-mediated mechanism that enables selective cleavage of four carbon-fluorine bonds while simultaneously constructing the furan ring system. This process begins with base-induced homolytic cleavage of the peroxide bond in polyfluorinated peroxygen compounds, generating radical species that undergo sequential transformations to form the critical carbon-carbon bonds required for furan ring formation. The organic sulfinate components serve dual roles as both radical acceptors and sulfone precursors, facilitating the construction of the unique bis-sulfone substituted furan architecture characteristic of these compounds. This mechanism operates through a carefully orchestrated series of radical addition and cyclization steps that proceed with high regioselectivity under mild thermal conditions, avoiding the need for external energy sources or specialized activation methods.
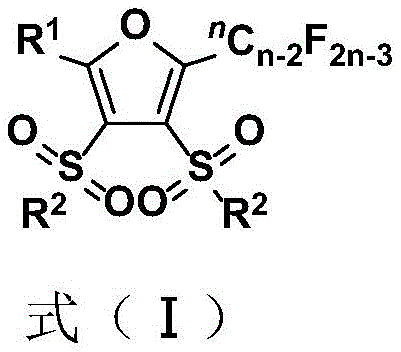
The strategic incorporation of multiple sulfone groups into the furan scaffold creates a unique electronic environment that enhances both stability and reactivity profiles compared to conventional furan derivatives. This structural feature also contributes significantly to the exceptional purity levels achieved through this synthetic route, as the polar sulfone moieties facilitate straightforward separation from non-polar impurities during purification. The mechanism demonstrates remarkable tolerance to various functional groups including halogens, nitriles, and carbonyl derivatives, enabling the synthesis of structurally diverse furan analogs without requiring protective group strategies. This inherent selectivity minimizes side reactions and by-product formation, resulting in cleaner reaction profiles that translate directly to higher yields and reduced downstream processing requirements.
The reaction pathway maintains excellent control over stereochemical outcomes through its radical-based mechanism, which avoids common issues associated with traditional acid-catalyzed cyclization methods that often lead to racemization or isomerization problems. This precise stereochemical control is particularly valuable for pharmaceutical applications where specific stereoisomers may exhibit dramatically different biological activities. The process achieves high atom economy by incorporating all components of the starting materials into the final product structure without generating significant waste streams. This green chemistry approach aligns perfectly with modern sustainability requirements while delivering superior product quality metrics essential for pharmaceutical intermediate manufacturing.
How to Synthesize Fluoroalkyl-Substituted Furan Compounds Efficiently
This patented synthesis route represents a significant advancement in the production of fluorinated heterocyclic compounds through its innovative three-component coupling strategy that operates under exceptionally mild conditions. The methodology leverages readily available starting materials and simple reaction parameters to achieve high-yielding transformations without requiring specialized equipment or hazardous reagents. This section provides a detailed overview of the standardized procedure developed from extensive optimization studies documented in the patent literature, highlighting critical parameters that ensure consistent product quality and process reliability at commercial scale. The following step-by-step guide outlines the precise implementation protocol that has been validated across multiple substrate classes to deliver optimal results.
- Combine polyfluorinated peroxygen compound with organic sulfinate in tert-butanol solvent under air atmosphere at 50°C with cesium carbonate as promoter.
- Stir the reaction mixture for 5-24 hours while monitoring progress via TLC analysis to ensure complete conversion of starting materials.
- Purify the reaction product through sequential water washing, ethyl acetate extraction, anhydrous sodium sulfate drying, and column chromatography separation.
Commercial Advantages for Procurement and Supply Chain Teams
This innovative synthesis methodology delivers substantial commercial benefits by addressing critical pain points in pharmaceutical intermediate manufacturing supply chains through its inherently robust and scalable process design. The elimination of transition metal catalysts removes a major source of supply chain vulnerability while simultaneously reducing quality control complexities associated with metal residue testing and removal procedures. The use of commercially available starting materials with stable supply chains ensures consistent raw material availability regardless of geopolitical factors that often disrupt specialized chemical markets. These advantages collectively enhance procurement flexibility while providing greater assurance of uninterrupted production schedules essential for meeting demanding pharmaceutical development timelines.
- Cost Reduction in Manufacturing: The process eliminates expensive transition metal catalysts and associated purification steps required to remove toxic metal residues, resulting in substantial cost savings through reduced raw material expenses and simplified downstream processing workflows. The mild reaction conditions minimize energy consumption while avoiding specialized equipment requirements that typically increase capital expenditures in traditional synthesis routes. The high functional group tolerance reduces the need for protective group strategies, further streamlining production processes and lowering overall manufacturing costs without compromising product quality or yield.
- Enhanced Supply Chain Reliability: By utilizing widely available starting materials with established global supply networks, this methodology significantly reduces dependency on specialized or single-source reagents that often create supply chain bottlenecks. The robust reaction profile maintains consistent performance across different batches of raw materials, minimizing quality variations that could disrupt production schedules. The simplified purification requirements reduce processing time and increase manufacturing throughput, enabling more responsive capacity adjustments to meet fluctuating market demands while maintaining stringent quality standards required for pharmaceutical applications.
- Scalability and Environmental Compliance: The process demonstrates exceptional scalability from laboratory to commercial production scales due to its straightforward reaction setup and minimal safety hazards associated with mild operating conditions. The elimination of toxic metal catalysts addresses critical environmental compliance concerns while reducing waste treatment costs associated with hazardous by-product disposal. The high atom economy and minimal by-product formation align with green chemistry principles, supporting sustainability initiatives without compromising economic viability or product quality requirements in pharmaceutical manufacturing environments.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial concerns regarding the implementation of this innovative synthesis methodology based on extensive validation studies and practical application experience documented in the patent literature. These answers reflect real-world insights gained from optimizing the process across diverse substrate classes while maintaining strict adherence to pharmaceutical quality standards required for commercial production environments.
Q: How does this novel synthesis method eliminate the need for transition metal catalysts?
A: The patented process utilizes a unique radical coupling mechanism between polyfluorinated peroxygen compounds and organic sulfinates, facilitated by simple inorganic bases like cesium carbonate, which eliminates the requirement for toxic transition metal catalysts while maintaining high selectivity and yield.
Q: What are the key advantages of this method for pharmaceutical intermediate production?
A: This method offers superior functional group tolerance, broad substrate universality, and avoids toxic metal residues in the final product, making it ideal for pharmaceutical applications where purity and safety are paramount.
Q: How does this process improve supply chain reliability for fluoroalkyl-substituted furan compounds?
A: By using readily available starting materials with simple reaction conditions that don't require specialized equipment or hazardous reagents, this process enables more consistent production with reduced supply chain vulnerabilities compared to traditional methods.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Fluoroalkyl-Substituted Furan Compound Supplier
Our company brings extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production of complex fluorinated heterocyclic compounds while maintaining stringent purity specifications through rigorous QC labs equipped with state-of-the-art analytical instrumentation. We have successfully implemented this patented methodology across multiple client projects, demonstrating consistent ability to deliver high-purity fluoroalkyl-substituted furan intermediates that meet exacting pharmaceutical industry requirements through our vertically integrated manufacturing platform. Our technical team combines deep expertise in radical chemistry with practical knowledge of regulatory compliance frameworks to ensure seamless technology transfer from laboratory development to commercial-scale manufacturing operations.
We invite you to request a Customized Cost-Saving Analysis from our technical procurement team to evaluate how this innovative synthesis route can optimize your specific manufacturing requirements while improving supply chain resilience. Please contact us to obtain specific COA data and route feasibility assessments tailored to your unique application needs within the pharmaceutical intermediate sector.