Scalable Synthesis of 5-Trifluoromethyl Imidazoles for Advanced Pharmaceutical Intermediates
Scalable Synthesis of 5-Trifluoromethyl Imidazoles for Advanced Pharmaceutical Intermediates
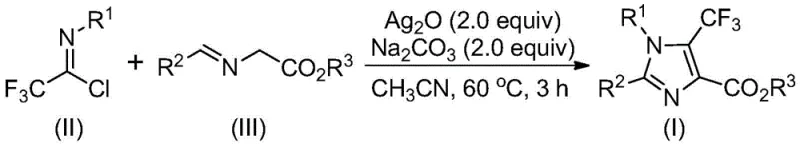
The pharmaceutical and fine chemical industries are constantly seeking robust, scalable pathways to access fluorinated heterocycles, which serve as critical scaffolds in modern drug discovery. Patent CN113735778A introduces a groundbreaking preparation method for 5-trifluoromethyl substituted imidazole compounds, addressing long-standing challenges in synthetic efficiency and cost-effectiveness. This technology leverages a transition metal silver oxide promoted [3+2] cycloaddition reaction, utilizing trifluoroethylimidoyl chloride and imidates as key building blocks. The significance of this innovation lies in its ability to produce fully substituted imidazole derivatives with exceptional yields, often reaching near-quantitative levels, under relatively mild thermal conditions ranging from 40°C to 80°C. For R&D directors and process chemists, this represents a pivotal shift away from complex, low-yielding legacy routes toward a streamlined protocol that ensures high purity and structural diversity.
Furthermore, the introduction of the trifluoromethyl group is strategically vital for enhancing the physicochemical properties of parent molecules, including improved metabolic stability, lipophilicity, and bioavailability. As evidenced by commercially successful drugs like metronidazole and losartan, the imidazole core is indispensable, and the addition of the CF3 moiety further amplifies its therapeutic potential. This patent not only validates the chemical feasibility of these transformations but also underscores the practical utility for generating diverse libraries of compounds for biological screening. By establishing a reliable foundation for synthesizing these high-value intermediates, the methodology supports the accelerated development of next-generation active pharmaceutical ingredients (APIs) and functional materials.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of trifluoromethyl-substituted imidazoles has been hindered by the reliance on expensive and difficult-to-handle synthons. Traditional literature methods primarily depend on the reaction of methylene amine ylides with trifluoromethyl-substituted imines via [3+2] cycloaddition. A major bottleneck in this conventional approach is the prerequisite synthesis of the trifluoromethyl-substituted imine, which necessitates the use of costly trifluoro-acetaldehyde ethyl hemiacetal compounds. This dependency creates significant supply chain vulnerabilities and inflates the overall cost of goods sold (COGS), making large-scale application economically unfeasible for many generic drug manufacturers. Additionally, the operational complexity associated with handling sensitive hemiacetals often leads to inconsistent batch-to-batch reproducibility and lower overall process mass intensity (PMI).
Beyond the economic drawbacks, the substrate scope of these older methods is frequently narrow, limiting the ability of medicinal chemists to explore diverse chemical space efficiently. The harsh conditions sometimes required to drive these reactions to completion can also lead to the formation of undesirable by-products and impurities, complicating downstream purification processes. For procurement managers, the reliance on specialized, high-cost reagents translates to longer lead times and increased inventory holding costs. Consequently, there has been a persistent industry demand for a more atom-economical and operationally simple alternative that utilizes commodity chemicals while maintaining high stereochemical and regiochemical control.
The Novel Approach
The novel approach detailed in the patent revolutionizes this landscape by employing cheap and easily obtainable trifluoroethylimidoyl chloride and imidates as the primary starting materials. This strategic selection of reagents eliminates the need for the prohibitively expensive hemiacetal precursors, fundamentally altering the cost structure of the synthesis. The reaction is facilitated by a promoter system consisting of silver oxide and an additive like sodium carbonate in an aprotic organic solvent, preferably acetonitrile. This catalytic system enables a highly efficient intermolecular carbon-carbon bond formation followed by intramolecular cyclization and oxidative aromatization. The result is a direct, one-pot transformation that delivers the target 5-trifluoromethyl imidazole with remarkable speed and efficiency.
Operational simplicity is another hallmark of this new methodology. The reaction proceeds smoothly at temperatures between 40°C and 80°C for just 2 to 4 hours, significantly reducing energy consumption compared to high-temperature reflux protocols. The work-up procedure is equally straightforward, involving simple filtration followed by standard column chromatography, which facilitates rapid isolation of the pure product. For supply chain heads, this translates to faster throughput times and reduced reactor occupancy, allowing for greater production capacity within existing infrastructure. The versatility of the method is further demonstrated by its tolerance to a wide range of functional groups, enabling the synthesis of diverse derivatives without the need for extensive protecting group strategies.
Mechanistic Insights into Silver Oxide Promoted Cyclization
The mechanistic pathway of this transformation offers fascinating insights into the role of the silver promoter in driving the reaction toward the desired aromatic product. The process initiates with an alkali-promoted intermolecular carbon-carbon bond formation between the trifluoroethylimidoyl chloride and the imidate, generating a bisimine intermediate. This step is crucial as it establishes the carbon framework necessary for the subsequent ring closure. Following this, an isomerization occurs, setting the stage for the silver-promoted intramolecular cyclization. The silver species acts as a Lewis acid, coordinating to the nitrogen atoms and facilitating the nucleophilic attack that closes the five-membered imidazoline ring. This coordination lowers the activation energy for cyclization, ensuring that the reaction proceeds rapidly even under mild thermal conditions.
The final and perhaps most critical step is the oxidative aromatization promoted by silver oxide. Unlike traditional dehydrogenation methods that might require stoichiometric oxidants or harsh conditions, the silver oxide in this system serves a dual purpose as both a promoter for cyclization and an oxidant for aromatization. This elegant cascade converts the 2-hydroimidazole intermediate directly into the stable, aromatic 5-trifluoromethyl-substituted imidazole. From an impurity control perspective, this mechanism is highly advantageous because it minimizes the accumulation of partially oxidized intermediates that could act as genotoxic impurities. The high selectivity of the silver-mediated process ensures that the final product profile is clean, reducing the burden on quality control laboratories and simplifying the regulatory filing process for new drug applications.
How to Synthesize 5-Trifluoromethyl Imidazole Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires careful attention to reagent ratios and solvent choice to maximize yield and purity. The patent specifies a preferred molar ratio of trifluoroethylimidoyl chloride to imidate to silver oxide of approximately 1:1.5:2, although a range of 1:1-2:1-3 is also effective. Acetonitrile is identified as the optimal solvent due to its ability to effectively dissolve the starting materials and promote the reaction kinetics, though tetrahydrofuran and dioxane are viable alternatives. The reaction time is optimized at 2 to 4 hours; extending beyond this window offers diminishing returns and may increase processing costs without significant gains in conversion. Detailed standardized operating procedures for scaling this reaction from milligram to kilogram quantities are essential for ensuring consistency.
- Combine trifluoroethylimidoyl chloride, imidate, silver oxide (promoter), and sodium carbonate (additive) in an organic solvent such as acetonitrile.
- Heat the reaction mixture to a temperature between 40°C and 80°C and maintain stirring for 2 to 4 hours to ensure complete conversion.
- Upon completion, filter the mixture, mix with silica gel, and perform column chromatography purification to isolate the high-purity 5-trifluoromethyl substituted imidazole product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain strategists, the adoption of this synthesis route offers compelling economic and logistical benefits that extend far beyond simple yield improvements. The primary driver of value is the substitution of exotic, high-cost reagents with commodity chemicals that are widely available in the global market. Trifluoroethylimidoyl chloride can be synthesized quickly from aromatic amines and trifluoroacetic acid, while imidates are derived from abundant aldehydes and glycines. This shift drastically reduces the raw material cost base and mitigates the risk of supply disruptions associated with single-source specialty reagents. Furthermore, the elimination of complex multi-step sequences in favor of a direct one-pot reaction significantly lowers the overall manufacturing overhead.
- Cost Reduction in Manufacturing: The economic impact of this process is profound due to the removal of expensive precursors and the simplification of the operational workflow. By utilizing silver oxide, which is relatively inexpensive compared to other transition metal catalysts like palladium or platinum, the direct cost of catalysis is minimized. Additionally, the high reaction efficiency means that less raw material is wasted in the form of by-products, leading to superior atom economy. The simplified post-treatment, which avoids complex extraction or distillation steps in favor of filtration and chromatography, further reduces labor and utility costs. These factors combine to create a manufacturing process that is inherently leaner and more cost-competitive, allowing for better margin protection in price-sensitive generic drug markets.
- Enhanced Supply Chain Reliability: Supply chain resilience is significantly bolstered by the use of robust, shelf-stable starting materials that do not require specialized cold-chain logistics or inert atmosphere storage to the same extent as sensitive organometallic reagents. The ability to source aldehydes, glycines, and aromatic amines from multiple global suppliers ensures that production schedules are not held hostage by vendor shortages. Moreover, the short reaction time of 2 to 4 hours allows for rapid turnover of reactor vessels, increasing the overall throughput capacity of the manufacturing facility. This agility enables suppliers to respond more quickly to fluctuating market demands and urgent customer orders, thereby strengthening the reliability of the supply partnership.
- Scalability and Environmental Compliance: From an environmental and scalability standpoint, this method aligns well with green chemistry principles by reducing solvent usage and waste generation. The high conversion rates mean that less solvent is required for purification, lowering the volume of hazardous waste that needs disposal. The use of acetonitrile, a common industrial solvent, facilitates easy recovery and recycling, further enhancing the sustainability profile of the process. As the patent notes, the method is expandable to gram-level and potentially industrial-scale production, indicating that the kinetics and heat transfer characteristics are favorable for larger reactors. This scalability ensures that the technology can grow with the product lifecycle, from early-stage clinical trials to commercial launch without the need for disruptive process re-engineering.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis technology. These answers are derived directly from the experimental data and technical specifications provided in the patent documentation, ensuring accuracy and relevance for process development teams. Understanding these nuances is critical for evaluating the feasibility of integrating this route into existing manufacturing pipelines.
Q: What are the key advantages of this synthesis method over conventional routes?
A: This method utilizes cheap and readily available starting materials like trifluoroethylimidoyl chloride and imidates, avoiding the expensive trifluoro-acetaldehyde ethyl hemiacetal used in traditional [3+2] cycloadditions. It offers extremely high reaction efficiency with yields approaching quantification for various substrates.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the patent explicitly states that the method can be expanded to gram-level reactions and provides possibilities for industrial large-scale production due to its simple operation, mild reaction conditions (40-80°C), and the use of inexpensive promoters like silver oxide.
Q: What is the substrate scope for this trifluoromethyl imidazole synthesis?
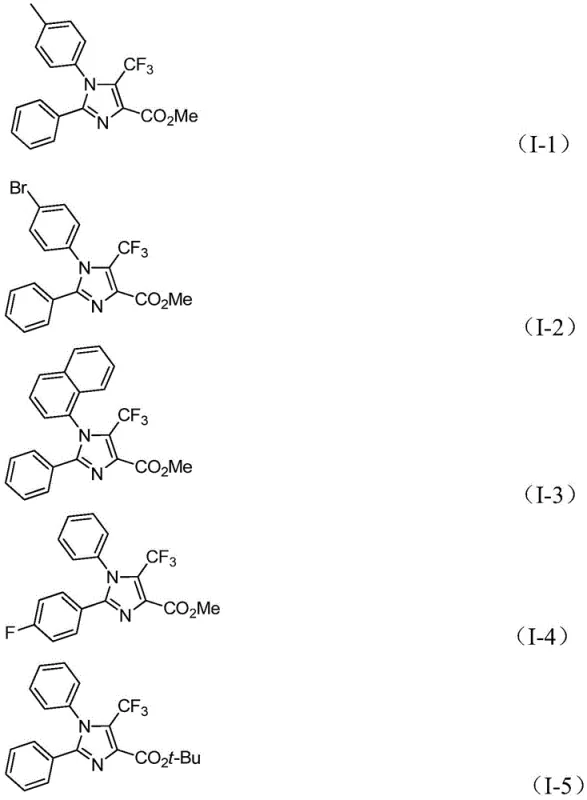
A: The method demonstrates strong substrate designability and wide tolerance for functional groups. It can synthesize diverse 1,2,4-substituted fully-substituted imidazole compounds where R1 and R2 can be various substituted aryl, naphthyl, or furyl groups, and R3 can be methyl, ethyl, or tert-butyl esters.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethyl Imidazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this silver-promoted cyclization technology for the production of high-value pharmaceutical intermediates. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from lab bench to market is seamless and efficient. Our state-of-the-art facilities are equipped to handle the specific requirements of fluorinated chemistry, including specialized containment and waste management systems. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of 5-trifluoromethyl imidazole meets the highest international standards for safety and efficacy.
We invite you to collaborate with us to leverage this innovative synthesis route for your next project. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. Please contact our technical procurement team today to request specific COA data for our catalog compounds or to discuss route feasibility assessments for your proprietary molecules. Together, we can accelerate the delivery of life-saving medicines to patients worldwide through superior chemical manufacturing excellence.