Scalable One-Step Asymmetric Synthesis of Chiral Tetrahydropyranones for Pharma Applications
Introduction to Advanced Asymmetric Catalysis
The landscape of chiral intermediate manufacturing is undergoing a significant transformation driven by the need for greener, more efficient synthetic routes. Patent CN109942553B introduces a groundbreaking methodology for the synthesis of chiral 3,4,6-trisubstituted tetrahydro-2H-pyran-2-one compounds, which are critical scaffolds in the development of bioactive pharmaceutical ingredients. This innovation leverages a one-step asymmetric catalytic reaction between readily available glycine derivatives and 3-substituted α,β-unsaturated aldehydes. By utilizing chiral secondary amine catalysts in conjunction with specific acidic additives, this process achieves high levels of stereocontrol without the reliance on precious transition metals. For R&D directors and procurement specialists, this represents a pivotal shift towards cost-effective and environmentally compliant manufacturing strategies that do not compromise on the stringent purity specifications required for global regulatory approval.
The Limitations of Conventional Methods vs. The Novel Approach
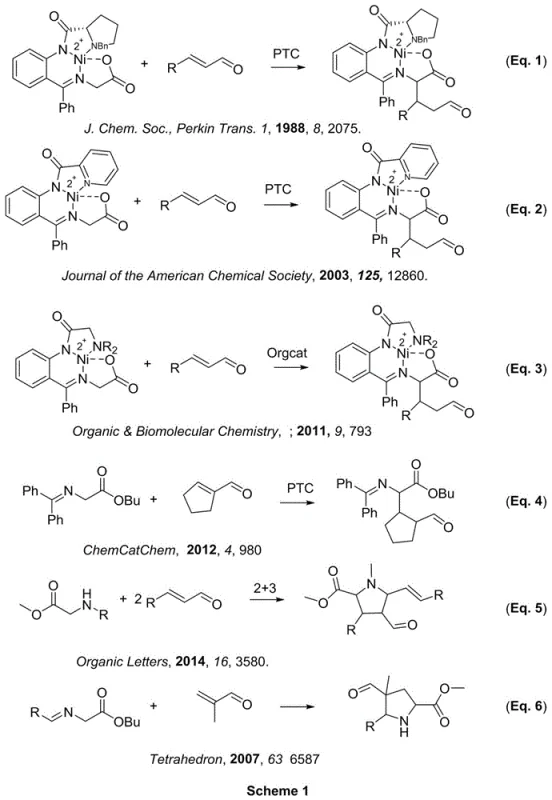
The Limitations of Conventional Methods
Historically, the construction of complex chiral amino acid derivatives and related heterocycles has relied heavily on transition metal catalysis, particularly nickel complexes, or phase-transfer catalysis systems. As illustrated in the background art, traditional approaches often involve the activation of the α-position of glycine imines through nickel coordination, requiring sophisticated chiral ligands and strict anhydrous conditions. These legacy methods, depicted in earlier literature schemes, frequently suffer from high operational complexity, sensitivity to moisture and oxygen, and the inherent challenge of removing trace heavy metal residues from the final API intermediate. Furthermore, multi-step sequences involving imine formation and subsequent cyclization increase the overall process mass intensity (PMI) and extend lead times, creating bottlenecks in the supply chain for high-purity pharmaceutical intermediates.
The Novel Approach
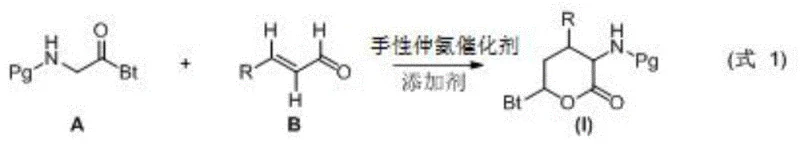
In stark contrast, the novel methodology disclosed in CN109942553B streamlines the synthesis into a single pot operation using organocatalysis. The core innovation involves the direct reaction of a glycine derivative bearing a specific protecting group (such as a perfluorocarboxylic acid amide) with an α,β-unsaturated aldehyde. This approach bypasses the need for metal coordination entirely, utilizing a chiral secondary amine catalyst, such as α,α-diphenylprolinol trimethylsilyl ether, to induce asymmetry. The reaction proceeds under mild conditions, typically between -10°C and 25°C, in common organic solvents like acetone or dichloromethane. This simplification not only enhances the safety profile of the manufacturing process but also drastically reduces the cost of goods sold (COGS) by eliminating expensive metal catalysts and complex workup procedures associated with metal scavenging.
Mechanistic Insights into Organocatalytic Cyclization
The success of this transformation lies in the precise activation modes enabled by the chiral secondary amine catalyst. The mechanism likely proceeds through the formation of a chiral enamine intermediate from the glycine derivative, which then undergoes a stereoselective Michael addition to the α,β-unsaturated aldehyde activated via iminium ion formation. The presence of an acidic additive, such as p-toluic acid or benzoic acid, plays a crucial role in facilitating proton transfer steps and stabilizing the transition state, thereby ensuring high diastereo- and enantioselectivity. Experimental data from the patent indicates that the choice of additive significantly impacts the yield and ee value, with p-toluic acid demonstrating superior performance in optimizing the reaction trajectory. This fine-tuning of the catalytic cycle allows for the construction of three contiguous stereocenters in a single operation, a feat that is challenging to achieve with conventional stepwise syntheses.
Furthermore, the structural integrity of the glycine derivative is maintained through the use of robust protecting groups like the benzotriazolyl (Bt) group or perfluoroacyl groups, which prevent racemization during the reaction. The resulting chiral 3,4,6-trisubstituted tetrahydro-2H-pyran-2-ones possess a rigid scaffold that locks the stereochemistry, preventing epimerization during downstream processing. This mechanistic robustness is vital for industrial scale-up, as it ensures consistent product quality across different batches. The ability to tolerate a wide range of substituents on the aldehyde component, including electron-rich and electron-deficient aromatic rings, underscores the versatility of this catalytic system in generating diverse chemical libraries for drug discovery programs.
How to Synthesize Chiral Tetrahydropyranones Efficiently
The practical implementation of this synthesis route is designed for ease of adoption in both laboratory and pilot plant settings. The protocol requires minimal specialized equipment, relying on standard stirring and temperature control units. The reaction mixture is typically monitored by thin-layer chromatography (TLC) to determine the endpoint, ensuring that the starting aldehyde is fully consumed before workup. Following the reaction, the crude product can be purified using standard column chromatography or crystallization techniques, yielding the target compound with high optical purity. For detailed operational parameters, stoichiometry, and specific purification protocols tailored to your specific substrate, please refer to the standardized synthesis guide below.
- Mix chiral secondary amine catalyst (0.1-50 mol%) and acid additive (0.1-50 mol%) in a suitable solvent like acetone and stir for 30 minutes.
- Add 1 equivalent of 3-substituted alpha,beta-unsaturated aldehyde and 1.2 equivalents of glycine derivative to the reaction mixture.
- Stir the reaction at -10 to 25°C until TLC indicates consumption of the aldehyde, then purify the crude mixture via column chromatography or crystallization.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this organocatalytic route offers compelling advantages that directly address the pain points of modern pharmaceutical supply chains. The shift from metal-based catalysis to organocatalysis fundamentally alters the cost structure and risk profile of producing these complex intermediates. By removing the dependency on volatile metal prices and the regulatory burden of heavy metal limits, manufacturers can achieve more predictable pricing and faster regulatory filings. The simplicity of the one-pot process also translates to reduced manufacturing cycles, allowing for quicker response times to market demands and improved inventory turnover rates for critical building blocks.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts, such as nickel, removes the necessity for expensive ligand systems and rigorous heavy metal clearance protocols, thereby significantly optimizing the cost structure. Additionally, the use of commodity solvents like acetone and commercially available glycine derivatives reduces raw material expenses, while the high atom economy of the one-step cyclization minimizes waste disposal costs associated with multi-step syntheses.
- Enhanced Supply Chain Reliability: The reliance on stable, shelf-stable organocatalysts and readily available aldehyde precursors mitigates the risk of supply disruptions often associated with specialized metal reagents. The robustness of the reaction conditions, which tolerate ambient temperatures and common solvents, ensures consistent production output regardless of minor fluctuations in facility utilities, securing a steady flow of high-purity intermediates for downstream API synthesis.
- Scalability and Environmental Compliance: The absence of toxic heavy metals simplifies the environmental, health, and safety (EHS) profile of the process, facilitating easier permitting and scale-up to commercial tonnage. The reduced generation of hazardous waste streams aligns with green chemistry principles, supporting corporate sustainability goals and reducing the environmental footprint of pharmaceutical manufacturing operations.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating the feasibility of this technology for their specific projects, we have compiled answers to common inquiries regarding the scope and application of this patent. These insights are derived directly from the experimental data and technical specifications outlined in the intellectual property documentation, providing a clear picture of the method's capabilities and limitations in real-world scenarios.
Q: What are the advantages of this organocatalytic method over traditional metal-catalyzed routes?
A: This method eliminates the need for expensive and toxic transition metal catalysts like nickel, simplifying the purification process by removing heavy metal clearance steps and reducing overall production costs while maintaining high enantioselectivity (up to 98% ee).
Q: What is the substrate scope for the alpha,beta-unsaturated aldehydes in this reaction?
A: The reaction demonstrates broad compatibility with various 3-substituted alpha,beta-unsaturated aldehydes, including those with aromatic groups (phenyl, naphthyl), heteroaromatic rings (furan, thiophene), and electron-withdrawing or donating substituents.
Q: Can the resulting tetrahydropyranone products be further derivatized?
A: Yes, the synthesized chiral tetrahydropyranones can be chemically transformed into other valuable scaffolds, such as chiral tetrahydropyranones with different protecting groups or chiral piperidones, expanding their utility in drug discovery.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral Tetrahydropyranone Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic value of adopting innovative synthetic routes like the one described in CN109942553B to enhance your product pipeline. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from bench-scale discovery to full-scale manufacturing is seamless and efficient. Our facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of chiral intermediate delivered meets the highest international standards for safety and efficacy.
We invite you to engage with our technical procurement team to discuss how this advanced organocatalytic technology can be integrated into your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the potential economic benefits specific to your volume requirements. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing us to demonstrate our commitment to being your trusted partner in the synthesis of complex pharmaceutical intermediates.