Scalable Synthesis of cis-Chiral-3-Fluoro-4-Hydroxypiperidine for Global Pharma Supply Chains
The pharmaceutical industry's relentless pursuit of metabolic stability and binding affinity has placed fluorinated heterocycles at the forefront of modern drug design. Patent CN115716799A introduces a transformative methodology for the preparation of cis-chiral-3-fluoro-4-hydroxypiperidine and its derivatives, addressing critical bottlenecks in the synthesis of these high-value scaffolds. By leveraging organoborohydride metal reagents, this technology enables the direct reduction of chiral fluorocarbonyl or dihydroxy precursors while rigorously maintaining the stereochemical integrity of the fluorine-bearing carbon center. For R&D Directors and Supply Chain Heads, this represents a paradigm shift from labor-intensive chromatographic separations to a robust, chemically driven stereoselective process. The ability to secure high-purity intermediates without relying on complex physical separation techniques fundamentally alters the cost structure and scalability potential for ErbB family receptor inhibitors and similar oncology therapeutics.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the acquisition of cis-chiral-3-fluoro-4-hydroxypiperidine structures has been heavily reliant on chiral chromatographic resolution, a technique fraught with economic and operational inefficiencies for large-scale manufacturing. While chromatography offers high selectivity in analytical settings, its application in industrial production is severely constrained by the high cost of chiral stationary phases and the limited throughput of separation equipment. Procurement Managers often face exorbitant pricing for kilogram-scale quantities produced via this route, as the process consumes significant solvent volumes and requires specialized infrastructure that is not universally available in standard chemical plants. Furthermore, the inherent batch-to-batch variability associated with column performance can lead to inconsistent supply continuity, posing a significant risk to downstream drug development timelines. The inability to easily scale chromatographic processes beyond pilot stages creates a formidable barrier for projects transitioning from clinical trials to commercial launch, often necessitating costly route redesigns late in the development cycle.
The Novel Approach
In stark contrast, the methodology disclosed in patent CN115716799A utilizes a chemical reduction strategy that inherently favors the formation of the desired cis-isomer through precise reagent control rather than physical separation. By employing specific organoborohydride metal reagents, such as lithium triethylborohydride or lithium tri-sec-butylborohydride, the reaction achieves a cis-selectivity greater than 97%, effectively rendering the product single-isomer without the need for chiral columns. This chemical approach simplifies the workflow dramatically, allowing for standard aqueous workups and crystallization or slurry purification techniques that are commonplace in multi-ton manufacturing facilities. The transition from a physical separation bottleneck to a chemical selectivity solution unlocks substantial capacity for production, enabling suppliers to meet surging demand for fluorinated piperidine intermediates with greater agility. This innovation not only reduces the direct cost of goods sold but also mitigates the supply chain risks associated with specialized separation technologies, ensuring a more reliable pipeline for critical pharmaceutical intermediates.
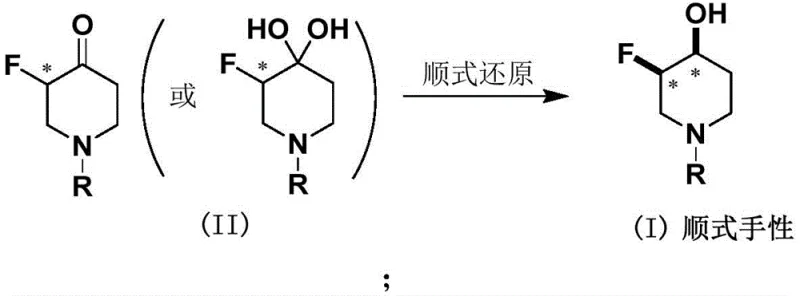
Mechanistic Insights into Organoborohydride-Mediated Stereoselective Reduction
The core of this technological advancement lies in the nuanced interaction between the bulky organoborohydride species and the sterically demanding fluorinated piperidone substrate. The reduction mechanism proceeds via a concerted hydride transfer where the steric bulk of the alkyl groups on the boron atom directs the approach of the hydride ion to the carbonyl face opposite to the adjacent fluorine substituent. This stereoelectronic control is crucial for maintaining the configuration of the chiral carbon atom where the fluorine atom is located, preventing epimerization which would otherwise compromise the biological activity of the final drug molecule. For R&D teams, understanding this mechanism validates the robustness of the process, as the selectivity is driven by fundamental molecular interactions rather than transient chromatographic conditions. The use of anhydrous tetrahydrofuran as the solvent further stabilizes the reactive borohydride species, ensuring a consistent kinetic profile that is essential for reproducible outcomes across different batch sizes. This level of mechanistic certainty provides a solid foundation for process validation and regulatory filing, reducing the technical risk associated with adopting new synthetic routes for GMP manufacturing.
Impurity control is another critical dimension where this chemistry excels, particularly regarding the suppression of trans-isomers and over-reduced byproducts. The high cis-selectivity (>97%) reported in the patent data indicates that the reaction pathway strongly disfavors the formation of the thermodynamic trans-product, likely due to the specific transition state geometry enforced by the reagent. This intrinsic purity profile minimizes the burden on downstream purification steps, allowing for simple slurry operations in solvent mixtures like n-hexane and ethyl acetate to achieve final specifications. From a quality assurance perspective, the absence of difficult-to-remove geometric isomers simplifies the impurity spectrum, making it easier to establish tight control strategies for commercial production. The preservation of the fluorine-carbon bond integrity throughout the reduction also ensures that no defluorinated side products are generated, which is a common concern in nucleophilic reactions involving fluorinated substrates. This clean reaction profile translates directly into higher overall yields and reduced waste generation, aligning with both economic and environmental sustainability goals.
How to Synthesize cis-Chiral-3-Fluoro-4-Hydroxypiperidine Efficiently
Implementing this synthesis route requires careful attention to reaction conditions, specifically temperature control and reagent stoichiometry, to maximize the benefits of the organoborohydride system. The process typically begins with the dissolution of the chiral fluorocarbonyl starting material in anhydrous tetrahydrofuran, followed by cooling to low temperatures (e.g., -60°C) to manage the exotherm and enhance stereocontrol. The subsequent addition of the reducing agent, such as lithium triethylborohydride, must be performed slowly under an inert atmosphere to prevent moisture degradation of the sensitive hydride species. Following the reaction completion, a standard quench with saturated ammonium chloride allows for the safe decomposition of excess boron species, facilitating a clean phase separation. The detailed standardized synthesis steps, including specific molar ratios and workup parameters validated for commercial scale-up, are outlined in the guide below.
- Dissolve the chiral fluorocarbonyl or dihydroxy starting material in anhydrous tetrahydrofuran under inert atmosphere.
- Cool the reaction mixture to low temperatures (e.g., -60°C) and slowly add the organoborohydride reducing agent.
- Allow the reaction to warm to room temperature, quench with saturated ammonium chloride, and purify via crystallization.
Commercial Advantages for Procurement and Supply Chain Teams
For Procurement Managers and Supply Chain Heads, the adoption of this organoborohydride reduction technology offers a compelling value proposition centered on cost efficiency and operational reliability. By eliminating the dependency on chiral chromatography, manufacturers can drastically reduce the capital expenditure required for specialized equipment and the ongoing operational costs associated with expensive chiral resins and solvents. This shift enables a more lean manufacturing model where the focus is on chemical conversion efficiency rather than physical separation capacity, leading to significant cost reduction in pharmaceutical intermediates manufacturing. The simplified workup procedure, involving basic extraction and crystallization, reduces the processing time per batch, thereby increasing the overall throughput of existing production assets without the need for major infrastructure upgrades. These efficiencies collectively contribute to a more competitive pricing structure for the final intermediate, allowing downstream drug developers to optimize their own cost of goods.
- Cost Reduction in Manufacturing: The elimination of chiral chromatography removes one of the most expensive unit operations in fine chemical synthesis, directly lowering the variable cost per kilogram of the product. Without the need for costly chiral stationary phases and the large volumes of solvents required for column elution, the material cost profile is significantly optimized. Furthermore, the high selectivity of the reaction minimizes the loss of valuable chiral starting materials to unwanted isomers, improving the overall mass balance and atom economy of the process. This efficient utilization of raw materials ensures that the cost of goods sold remains stable even when scaling to multi-ton quantities, providing predictable budgeting for long-term supply agreements.
- Enhanced Supply Chain Reliability: Relying on standard chemical reduction rather than specialized separation equipment diversifies the potential supplier base, as more contract manufacturing organizations possess the capability to perform hydride reductions. This broader manufacturing base reduces the risk of supply disruption caused by equipment downtime or capacity constraints at a single specialized facility. The robustness of the reaction conditions, which tolerate standard industrial handling procedures, ensures consistent batch quality and delivery timelines. Consequently, pharmaceutical companies can secure a more resilient supply chain for high-purity fluorinated intermediates, mitigating the risks associated with single-source dependencies on niche chromatography service providers.
- Scalability and Environmental Compliance: The process is inherently designed for scale, utilizing reagents and solvents that are manageable in large-scale reactors with appropriate safety controls. The avoidance of complex waste streams associated with chiral column regeneration simplifies environmental compliance and waste treatment protocols. Additionally, the high atom economy and reduced solvent consumption align with green chemistry principles, lowering the environmental footprint of the manufacturing process. This scalability ensures that the supply can grow seamlessly from clinical trial requirements to commercial launch volumes, supporting the rapid market entry of new therapies without the typical delays associated with process re-engineering.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis method. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on the practical implications for industrial adoption. Understanding these details helps stakeholders evaluate the feasibility of integrating this route into their existing supply chains and development pipelines. The answers reflect the consensus on the technology's capability to deliver high-purity materials with improved efficiency compared to legacy methods.
Q: How does this method improve upon chiral chromatography?
A: Unlike chiral chromatography which is costly and difficult to scale, this organoborohydride reduction achieves high cis-selectivity (>97%) directly, eliminating the need for expensive separation equipment.
Q: Is the stereochemistry at the fluorine position maintained?
A: Yes, the patent data confirms that the configuration of the chiral carbon atom where the fluorine atom is located is strictly maintained throughout the reduction process.
Q: What are the typical purity levels achieved?
A: The method consistently yields products with purity exceeding 97% after simple slurry purification, making it suitable for stringent pharmaceutical applications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable cis-Chiral-3-Fluoro-4-Hydroxypiperidine Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of robust synthetic routes for complex fluorinated intermediates in the global pharmaceutical landscape. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from benchtop discovery to full-scale manufacturing. Our commitment to quality is underpinned by stringent purity specifications and rigorous QC labs that validate every batch against the highest industry standards. We understand that the consistency of chiral fluorinated building blocks is critical for the efficacy and safety of the final drug product, and our technical team is dedicated to maintaining the stereochemical integrity required for your specific application.
We invite you to collaborate with us to leverage this advanced organoborohydride reduction technology for your next project. Contact our technical procurement team today to request a Customized Cost-Saving Analysis that quantifies the potential efficiencies of switching to this scalable route. We are prepared to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can support your supply chain goals. Let us help you secure a reliable source of high-purity intermediates that drives both innovation and commercial success.