Scalable Synthesis of 5-Trifluoromethyl Imidazoles for Advanced Pharmaceutical Manufacturing
Scalable Synthesis of 5-Trifluoromethyl Imidazoles for Advanced Pharmaceutical Manufacturing
The integration of trifluoromethyl groups into heterocyclic scaffolds represents a cornerstone strategy in modern medicinal chemistry, profoundly enhancing the metabolic stability, lipophilicity, and bioavailability of drug candidates. As detailed in the recent patent disclosure CN113735778A, a breakthrough methodology has been established for the efficient preparation of 5-trifluoromethyl substituted imidazole compounds, addressing critical bottlenecks in the supply chain of high-value pharmaceutical intermediates. This novel synthetic route leverages a silver oxide-promoted [3+2] cycloaddition strategy, offering a robust alternative to legacy processes that have long plagued R&D teams with issues of cost and scalability. The significance of this technology cannot be overstated, as imidazole derivatives serve as the structural backbone for a vast array of clinically essential medications, ranging from antifungal agents like econazole to antihistamines and angiotensin II receptor blockers.
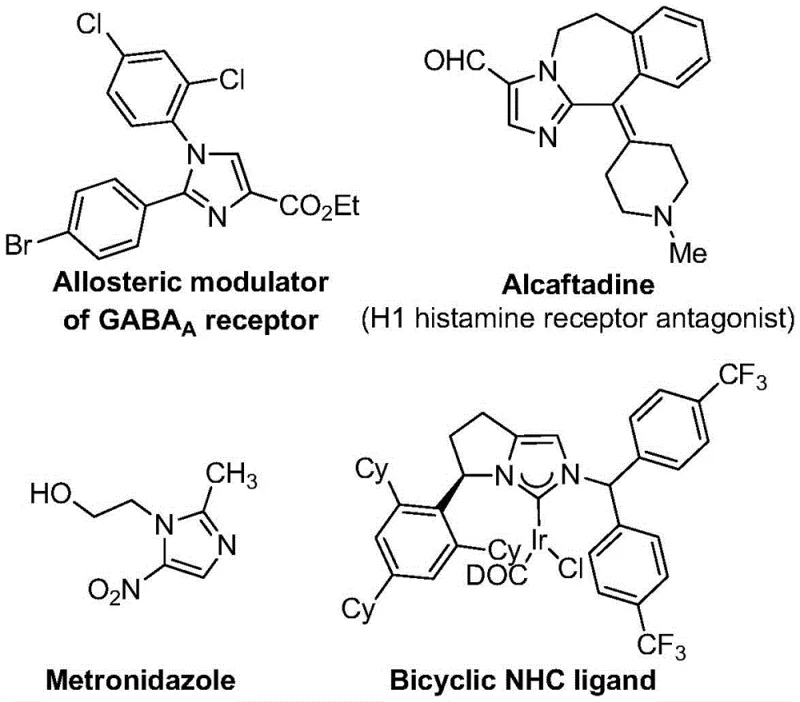
The versatility of the imidazole core is evident in its widespread application across diverse therapeutic areas, yet the introduction of the trifluoromethyl moiety at the 5-position has historically presented significant synthetic challenges. Traditional approaches often rely on complex multi-step sequences or unstable synthons that limit their utility in commercial settings. The methodology described in patent CN113735778A circumvents these limitations by utilizing trifluoroethylimidoyl chloride and imidates as primary building blocks. This strategic shift not only simplifies the operational workflow but also expands the chemical space accessible to process chemists, enabling the rapid generation of diverse libraries for structure-activity relationship (SAR) studies. For procurement managers and supply chain directors, this translates to a more reliable source of critical intermediates with reduced lead times and enhanced cost predictability.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of trifluoromethyl-substituted imidazoles has been dominated by [3+2] cycloaddition reactions involving methylene amine ylides and trifluoromethyl-substituted imines. While chemically sound, this conventional pathway suffers from severe economic and logistical drawbacks that hinder its adoption in large-scale manufacturing. The primary bottleneck lies in the preparation of the trifluoromethyl-substituted imine synthon, which necessitates the use of expensive trifluoro-acetaldehyde ethyl hemiacetal compounds. These precursors are not only costly but also exhibit limited stability and availability, creating a fragile supply chain vulnerable to market fluctuations. Furthermore, the scale-up of these traditional methods is often restricted by the handling difficulties associated with volatile or sensitive reagents, leading to inconsistent batch quality and lower overall throughput. For a reliable pharmaceutical intermediate supplier, relying on such inefficient routes compromises the ability to deliver consistent volumes required by global API manufacturers.
The Novel Approach
In stark contrast, the innovative process disclosed in patent CN113735778A utilizes trifluoroethylimidoyl chloride and imidates as the foundational substrates, driven by a transition metal silver oxide promotion system. This approach fundamentally alters the economic landscape of imidazole synthesis by replacing expensive, niche reagents with cheap and readily available starting materials. The reaction proceeds through a streamlined one-pot protocol where the promoter and additive facilitate the formation of the heterocyclic ring under mild thermal conditions. As illustrated in the general reaction scheme below, the convergence of these simple precursors allows for the direct construction of the fully substituted imidazole core with exceptional efficiency.
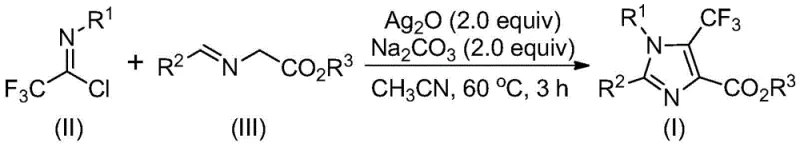
The operational simplicity of this novel method is a game-changer for process development teams. By eliminating the need for pre-formed unstable imines, the process reduces the number of unit operations and minimizes waste generation. The use of acetonitrile as a preferred solvent further enhances the practicality of the method, as it is a standard industrial solvent with well-established recovery and recycling protocols. This shift towards atom-economical and operationally simple chemistry aligns perfectly with the industry's drive towards green manufacturing principles, offering substantial cost reduction in API intermediate manufacturing while maintaining high standards of purity and yield.
Mechanistic Insights into Silver-Promoted Cyclization
Understanding the mechanistic underpinnings of this transformation is crucial for R&D directors aiming to optimize the process for specific substrates. The reaction initiates with an alkali-promoted intermolecular carbon-carbon bond formation between the trifluoroethylimidoyl chloride and the imidate, generating a reactive bisimine intermediate. This step is critical as it sets the stereochemical and regiochemical stage for the subsequent cyclization. Following the formation of the bisimine, the system undergoes an isomerization process that aligns the reactive centers for ring closure. The presence of silver oxide is pivotal at this juncture; it acts not merely as a base but as a Lewis acid promoter that activates the imine functionality towards nucleophilic attack, facilitating the intramolecular cyclization to form a 2-hydroimidazole species.
The final and perhaps most elegant step of the mechanism is the oxidative aromatization, also mediated by the silver species. This transformation converts the dihydro-intermediate into the fully aromatic 5-trifluoromethyl-substituted imidazole compound. The ability of silver oxide to drive this oxidation without the need for external stoichiometric oxidants (like peroxides or hypervalent iodine) is a significant safety and cost advantage. It ensures that the reaction profile remains clean, minimizing the formation of over-oxidized byproducts or polymeric tars that often complicate downstream purification. This mechanistic clarity allows for precise control over impurity profiles, ensuring that the resulting high-purity pharmaceutical intermediates meet the stringent specifications required for GMP production environments.
How to Synthesize 5-Trifluoromethyl Imidazole Efficiently
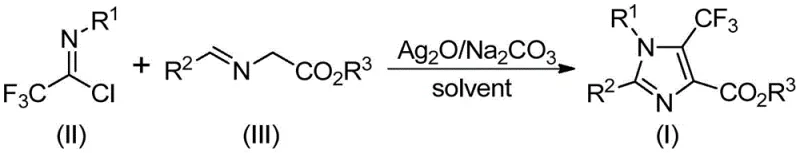
The execution of this synthesis is designed for robustness and reproducibility, making it ideal for technology transfer from the laboratory to the pilot plant. The standard protocol involves charging a reactor with the requisite molar equivalents of trifluoroethylimidoyl chloride, the specific imidate derivative, silver oxide, and sodium carbonate in acetonitrile. The mixture is then heated to a moderate temperature range of 40-80°C, typically optimized at 60°C, and stirred for a duration of 2 to 4 hours. Monitoring the reaction progress via TLC or HPLC ensures that the conversion is complete before proceeding to workup. The detailed standardized synthesis steps for specific derivatives are outlined in the guide below.
- Combine trifluoroethylimidoyl chloride, imidate, silver oxide (2.0 equiv), and sodium carbonate (2.0 equiv) in an organic solvent such as acetonitrile.
- Heat the reaction mixture to 60°C and stir for approximately 3 hours to ensure complete conversion.
- Filter the reaction mixture, mix with silica gel, and purify via column chromatography to isolate the target 5-trifluoromethyl imidazole compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers compelling strategic advantages that extend beyond mere chemical elegance. The primary value proposition lies in the drastic simplification of the raw material portfolio. By shifting away from specialized fluorinated hemiacetals to commodity chemicals like aromatic amines, aldehydes, and glycines, the supply chain becomes significantly more resilient. These starting materials are produced globally in massive volumes, insulating the manufacturing process from the volatility associated with niche fluorine chemistry suppliers. This diversification of the supply base is a critical risk mitigation strategy for ensuring continuous production of life-saving medications.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven by the elimination of expensive catalysts and the use of low-cost starting materials. Unlike palladium or rhodium-catalyzed cross-couplings that require expensive ligands and rigorous metal scavenging steps, this silver-promoted method utilizes relatively inexpensive silver oxide. Furthermore, the high reaction efficiency, with yields often approaching quantification, means that raw material waste is minimized. The simplified post-treatment process, involving basic filtration and standard chromatography, reduces the consumption of silica gel and solvents, leading to substantial cost savings in the overall cost of goods sold (COGS).
- Enhanced Supply Chain Reliability: The reliance on broadly available chemical feedstocks ensures that production schedules are not held hostage by the lead times of exotic reagents. The robustness of the reaction conditions—tolerating a wide range of functional groups including halogens, alkyls, and ethers—means that a single platform technology can be used to manufacture a diverse array of intermediates. This flexibility allows manufacturers to respond rapidly to changing market demands without the need for extensive process re-validation. The ability to scale from gram-level discovery batches to multi-kilogram commercial runs without significant re-optimization further strengthens supply continuity.
- Scalability and Environmental Compliance: From an environmental and safety perspective, the process is highly favorable. The use of acetonitrile, a solvent with established recovery infrastructure, combined with the absence of toxic heavy metals or explosive oxidants, simplifies waste management and regulatory compliance. The high atom economy of the [3+2] cycloaddition ensures that the majority of the mass of the starting materials ends up in the final product, reducing the E-factor (environmental factor) of the process. This alignment with green chemistry principles not only lowers disposal costs but also enhances the sustainability profile of the final pharmaceutical product, a key metric for modern ESG-conscious supply chains.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing a clear picture of the method's capabilities and limitations for potential partners.
Q: What are the key advantages of using trifluoroethylimidoyl chloride over traditional synthons?
A: Unlike traditional methods requiring expensive trifluoro-acetaldehyde ethyl hemiacetal, trifluoroethylimidoyl chloride is derived from readily available and inexpensive starting materials, significantly lowering raw material costs and improving scalability for industrial applications.
Q: How does the silver oxide promoter influence the reaction mechanism?
A: Silver oxide acts as a crucial promoter that facilitates the intramolecular cyclization of the bisimine intermediate and drives the final oxidative aromatization step, ensuring high yields of the fully substituted aromatic imidazole ring without the need for harsh oxidants.
Q: Is this synthesis method suitable for large-scale commercial production?
A: Yes, the method utilizes cheap and commercially available reagents, operates under mild conditions (60°C), and demonstrates high reaction efficiency with yields approaching quantification, making it highly amenable to kilogram-to-ton scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethyl Imidazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the synthetic methodologies described in patent CN113735778A for the next generation of fluorinated pharmaceuticals. As a premier CDMO partner, we possess the technical expertise and infrastructure to translate this academic innovation into commercial reality. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project moves seamlessly from the bench to the market. We are committed to delivering stringent purity specifications through our rigorous QC labs, guaranteeing that every batch of 5-trifluoromethyl imidazole intermediate meets the exacting standards of the global pharmaceutical industry.
We invite you to collaborate with us to leverage this cost-effective and scalable technology for your pipeline. By partnering with our technical procurement team, you can obtain a Customized Cost-Saving Analysis tailored to your specific molecule. We encourage you to reach out today to request specific COA data and comprehensive route feasibility assessments, allowing us to demonstrate how our advanced manufacturing capabilities can optimize your supply chain and accelerate your time to market.