Scalable Synthesis of Antineoplastic Drug Trepetidine Intermediate from L-Tyrosine
Scalable Synthesis of Antineoplastic Drug Trepetidine Intermediate from L-Tyrosine
The pharmaceutical industry continuously seeks robust and scalable pathways for complex antineoplastic agents, particularly for marine-derived drugs like Trabectedin (Yondelis). Patent CN111499525B discloses a groundbreaking synthetic method for producing the key chiral intermediate, (S)-3-methyl-4-methoxy-5-hydroxy phenylalaninol (Compound 2), which is essential for the total synthesis of Trabectedin. This innovation addresses critical bottlenecks in the supply chain by utilizing a cheap natural amino acid, L-tyrosine derivative, as the starting chiral pool material. Unlike previous methods that relied on expensive asymmetric catalysis, this novel approach leverages conventional transformation reactions including benzyl protection, selective phenol hydroxylation, and Suzuki coupling to achieve high chiral purity. For R&D directors and procurement managers, this represents a significant shift towards more sustainable and cost-efficient manufacturing of high-value pharmaceutical intermediates.
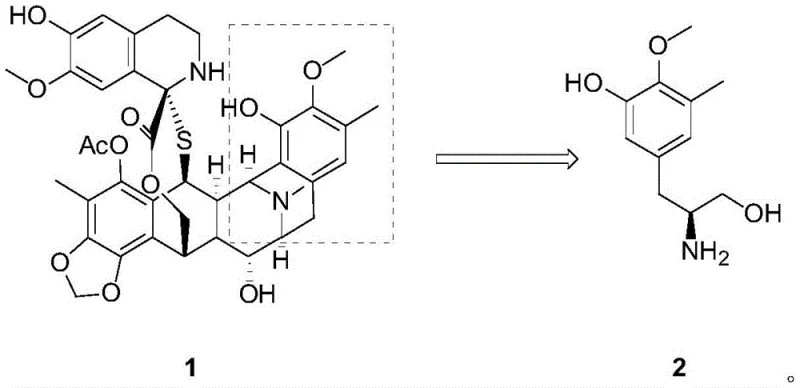
The strategic importance of Compound 2 cannot be overstated, as it serves as the foundational building block for the complex tetrahydroisoquinoline core of Trabectedin. The patent outlines a six-step sequence that transforms a readily available L-tyrosine derivative into the target molecule with exceptional stereochemical control. By avoiding the pitfalls of cryogenic chemistry and exotic reagents, this method offers a reliable pathway for commercial scale-up of complex pharmaceutical intermediates. The process ensures that the chiral center, originally derived from the natural amino acid, is preserved with 99% enantiomeric excess (ee) throughout the synthesis, guaranteeing the quality required for downstream API production.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this invention, the synthesis of Compound 2 was heavily reliant on the methodology reported by the E.J. Corey group, which presented substantial hurdles for industrial adoption. As illustrated in the prior art scheme, the construction of the chiral amino acid ester required the use of a sophisticated and prohibitively expensive chiral phase transfer catalyst, specifically O-allyl-N-(9-anthracenemethyl) cinchona alkaloid. Furthermore, the reaction conditions were extremely demanding, necessitating precise temperature control at -78°C to maintain any degree of stereoselectivity. Even under these rigorous conditions, the resulting chiral purity was only approximately 90% ee, which is often insufficient for modern regulatory standards without additional, yield-lossing purification steps. These factors combined to create a process with high operational costs and significant scalability risks.
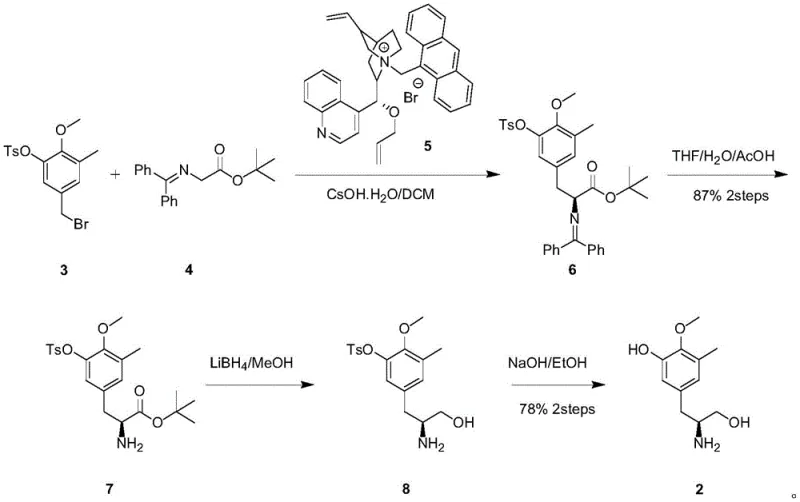
The Novel Approach
In stark contrast, the method disclosed in CN111499525B utilizes a chiral pool strategy starting from an inexpensive L-tyrosine derivative, effectively bypassing the need for asymmetric induction during the bond-forming steps. The overall synthetic route, depicted below, demonstrates a logical progression of functional group manipulations that are well-suited for large-scale reactors. By installing bromine handles early in the sequence, the inventors enabled a highly selective methylation via Suzuki coupling, a reaction known for its robustness and tolerance of various functional groups. This approach not only simplifies the operation but also drastically improves the economic profile of the synthesis by replacing rare catalysts with abundant natural materials and common transition metal catalysts like palladium.
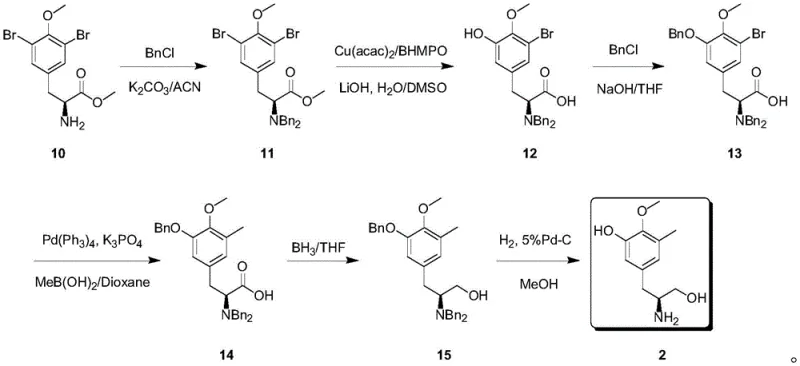
Mechanistic Insights into Suzuki Coupling and Selective Hydroxylation
A critical step in this synthesis is the palladium-catalyzed methylation (Step D), where the aryl bromide moiety in Compound 13 is converted to the corresponding methyl derivative (Compound 14). This transformation utilizes methylboronic acid and a palladium tetrakis(triphenylphosphine) catalyst in the presence of potassium phosphate. The mechanism involves the oxidative addition of the aryl bromide to the Pd(0) species, followed by transmetallation with the organoboron reagent and subsequent reductive elimination to forge the carbon-carbon bond. This step is pivotal because it installs the crucial methyl group at the 3-position of the aromatic ring with high regioselectivity, dictated by the positioning of the bromine atom introduced in earlier steps. The use of mild bases and temperatures around 90-100°C ensures that the sensitive ester and ether functionalities remain intact.
Equally important is the selective phenol hydroxylation (Step B), which converts the 3,5-dibromo intermediate 11 into the 3-bromo-5-hydroxy species 12. This reaction employs a copper catalytic system with Cu(acac)2 and a specific ligand, BHMPPO, in a DMSO/water mixture. The mechanism likely involves a copper-mediated nucleophilic aromatic substitution or oxidation process that selectively targets one of the bromine atoms ortho to the methoxy group. This selectivity is paramount; retaining one bromine atom allows for the subsequent Suzuki coupling, while revealing the hydroxyl group provides the necessary handle for the final structural motif of the trabectedin side chain. The ability to differentiate between two chemically similar bromine atoms showcases the precision of this synthetic design.
How to Synthesize (S)-3-methyl-4-methoxy-5-hydroxy phenylalaninol Efficiently
The synthesis of this high-value intermediate is designed for operational simplicity, utilizing standard unit operations common in fine chemical manufacturing. The process begins with the N-protection of the tyrosine derivative, followed by the critical copper-catalyzed hydroxylation and subsequent protection steps. The detailed standardized synthetic protocol, including specific reagent ratios, temperature profiles, and workup procedures for each of the six steps, is outlined below to assist process chemists in replicating this high-yielding route.
- Protect the amino group of L-tyrosine derivative 10 with benzyl chloride to form dibenzyl ester 11.
- Perform selective phenol hydroxylation on compound 11 using copper catalysis to obtain phenol 12.
- Protect the new phenolic hydroxyl group with benzyl chloride to yield compound 13.
- Execute Suzuki methylation using methyl boronic acid to install the methyl group, forming compound 14.
- Reduce the ester functionality using borane to generate the amino alcohol 15.
- Final hydrogenation debenzylation over Pd-C to afford the target intermediate 2.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this new synthetic route offers compelling advantages that directly impact the bottom line and supply security. The primary driver for cost reduction is the elimination of the expensive chiral phase transfer catalysts and the associated cryogenic infrastructure required by the legacy Corey method. By shifting to a chiral pool starting material, the process removes a major variable cost component and simplifies the reactor requirements, allowing for production in standard glass-lined or stainless steel vessels without the need for specialized low-temperature cooling systems. This fundamental change in the process architecture leads to substantial cost savings in utility consumption and capital expenditure.
- Cost Reduction in Manufacturing: The replacement of exotic chiral catalysts with commodity chemicals like benzyl chloride and methylboronic acid significantly lowers the raw material bill of materials. Furthermore, the high yields observed in key steps, such as the 96% yield in the benzyl protection step and 97% in the final hydrogenation, minimize waste generation and maximize throughput. The avoidance of cryogenic conditions (-78°C) also results in drastic energy savings, as maintaining such low temperatures on a multi-kilogram or ton scale is energetically intensive and costly. These factors combine to create a much more economically viable process for cost reduction in API manufacturing.
- Enhanced Supply Chain Reliability: Sourcing strategies are greatly improved by the use of L-tyrosine derivatives, which are abundant, renewable, and commercially available from multiple global suppliers. This reduces the risk of supply disruption associated with single-source specialty reagents like cinchona alkaloid derivatives. Additionally, the robustness of the Suzuki coupling and hydrogenation steps means that the process is less susceptible to batch failures due to minor fluctuations in reaction parameters. This reliability ensures a consistent flow of high-purity pharmaceutical intermediates to downstream customers, stabilizing the entire production schedule.
- Scalability and Environmental Compliance: The synthetic route is inherently scalable, utilizing reactions that are well-understood in industrial settings. The use of aqueous workups and standard organic solvents facilitates efficient solvent recovery and recycling, aligning with green chemistry principles. The high chiral purity (99% ee) achieved without chromatographic purification reduces the solvent load associated with purification, thereby lowering the environmental footprint. This makes the process not only easier to scale from pilot plant to commercial production but also more compliant with increasingly stringent environmental regulations regarding waste disposal and solvent emissions.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthetic method. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on the feasibility and advantages of adopting this technology for your supply chain.
Q: What is the chiral purity of the intermediate produced by this method?
A: The process maintains excellent stereochemical integrity throughout all six steps, consistently delivering the final product with 99% ee, which is superior to the 90% ee achieved by prior art methods.
Q: Why is this route considered more cost-effective than the Corey method?
A: This route eliminates the need for expensive chiral phase transfer catalysts like O-allyl-N-(9-anthracenemethyl) cinchona alkaloids and avoids cryogenic reaction conditions (-78°C), significantly reducing operational costs.
Q: Is this synthesis suitable for large-scale industrial production?
A: Yes, the use of robust reactions such as Suzuki coupling and standard hydrogenation, combined with mild temperature ranges (60-100°C), makes this process highly amenable to commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trepetidine Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the development of life-saving oncology drugs. Our team of expert process chemists has extensively evaluated the route disclosed in CN111499525B and confirmed its potential for seamless technology transfer. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can move rapidly from bench scale to full manufacturing. Our facilities are equipped with state-of-the-art rigorous QC labs capable of verifying the stringent purity specifications required for antineoplastic intermediates, including chiral HPLC analysis to guarantee the 99% ee benchmark.
We invite you to collaborate with us to optimize this synthesis further or to integrate it into your existing supply chain. Our technical sales team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Please contact our technical procurement team today to request specific COA data and discuss route feasibility assessments for your next project. Let us be your partner in delivering reliable, cost-effective, and high-purity solutions for the global pharmaceutical market.