Revolutionizing Isoxazole Synthesis: A Scalable One-Pot Strategy for High-Purity Pharmaceutical Intermediates
Introduction to Advanced Isoxazole Manufacturing
The strategic incorporation of fluorine atoms into heterocyclic scaffolds remains a cornerstone of modern medicinal chemistry, particularly for enhancing metabolic stability and membrane permeability in drug candidates. Patent CN108069918B introduces a groundbreaking one-pot methodology for the preparation of 3-difluoromethyl-substituted isoxazole compounds, addressing long-standing inefficiencies in synthetic organic chemistry. This innovation leverages readily available difluoroethylamine as a difluoromethyl source, coupling it directly with terminal alkynes under the catalysis of inexpensive copper salts. For R&D directors and procurement specialists seeking a reliable pharmaceutical intermediate supplier, this technology represents a paradigm shift towards greener, more cost-effective manufacturing processes that do not compromise on purity or structural diversity.
The significance of this patent lies in its ability to bypass the tedious multi-step sequences traditionally required to install the difluoromethyl group at the 3-position of the isoxazole ring. By streamlining the synthesis into a single operational unit, the method drastically reduces solvent consumption, energy usage, and overall processing time. This aligns perfectly with the industry's growing demand for sustainable chemical manufacturing and rapid lead optimization. As we delve deeper into the technical specifics, it becomes evident that this approach offers a robust platform for the commercial scale-up of complex pharmaceutical intermediates, ensuring a stable supply chain for critical drug development projects.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 3-difluoromethyl isoxazoles has been plagued by significant operational hurdles and economic inefficiencies. Early methodologies, such as the one reported by Linderman in 1989, relied on the cyclization of difluoromethyl-substituted alkynones with hydroxylamine hydrochloride. However, the preparation of these alkynone precursors often involves harsh reaction conditions and suffers from low overall yields. Furthermore, the final cyclization step frequently exhibits poor regioselectivity, leading to mixtures of N-O and O-N isomers that are difficult to separate, thereby complicating downstream purification and increasing production costs.
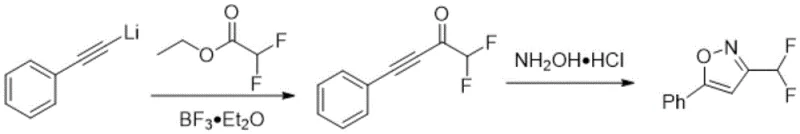
Subsequent attempts to improve this chemistry, such as the 2015 method by Schmitt and Leroux, introduced fluoroalkylamine reagents converted into fluoroamine salts. While this offered a different mechanistic pathway, it necessitated the pre-preparation of enol silyl ethers, adding extra synthetic steps and generating silicon-based waste. The requirement for specialized reagents like boron trifluoride diethyl etherate further escalates the safety risks and handling costs associated with the process. Similarly, the 2017 approach by Mykhaliuk utilized ethyl difluoroacetate reduced to difluoroacetaldehyde, followed by oxidation with N-chlorosuccinimide (NCS). This route is not only lengthy but also employs hazardous oxidants and strong reducing agents like lithium aluminium hydride, posing significant challenges for safe industrial implementation.
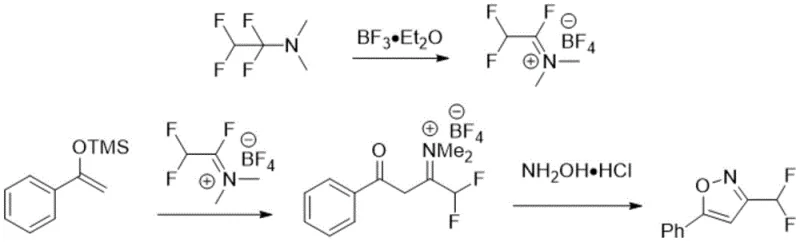
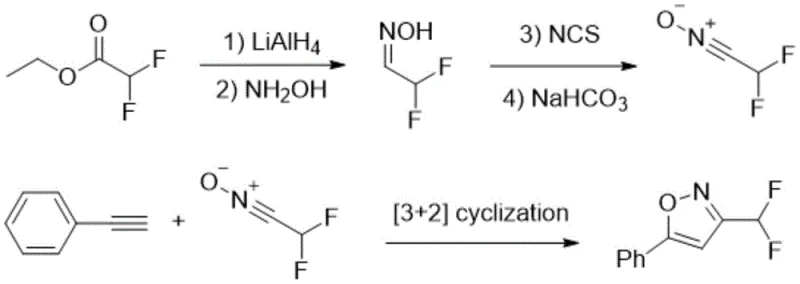
The Novel Approach
In stark contrast to these legacy methods, the invention disclosed in CN108069918B proposes a direct, one-pot coupling strategy that fundamentally simplifies the synthetic landscape. The core of this innovation is the in situ generation of fluorodiazomethane from commercially available difluoroethylamine, which then undergoes a copper-catalyzed coupling with terminal alkynes. This eliminates the need for isolating unstable or hazardous intermediates, thereby enhancing process safety and operational simplicity. The reaction proceeds under mild conditions, typically at room temperature, which significantly lowers energy consumption compared to the cryogenic or high-temperature requirements of previous techniques.
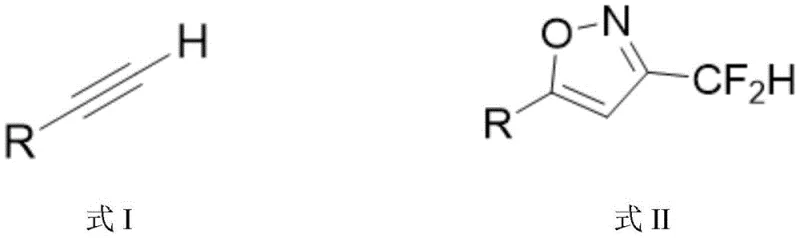
Moreover, this novel approach demonstrates exceptional functional group tolerance, accommodating a wide array of substituents on the alkyne substrate without the need for protecting groups. Whether the substrate contains electron-withdrawing nitro groups or electron-donating methoxy groups, the reaction maintains high efficiency and selectivity. This versatility is crucial for medicinal chemists who need to rapidly explore structure-activity relationships (SAR) by synthesizing diverse libraries of isoxazole derivatives. The use of cheap and abundant copper and zinc catalysts further underscores the economic viability of this method, making it an attractive option for cost reduction in API manufacturing on a commercial scale.
Mechanistic Insights into Copper-Catalyzed Cyclization
The mechanistic pathway of this transformation is believed to involve the generation of a reactive difluoromethyl ketoxime intermediate, which subsequently undergoes a [3+2] cycloaddition. Initially, the difluoroethylamine is activated by tert-butyl nitrite and acetic acid to form a diazo species in situ. This highly reactive intermediate then couples with the terminal alkyne in the presence of the copper(I) iodide and zinc bromide catalyst system. The zinc salt likely plays a crucial role in stabilizing the transition state or activating the alkyne towards nucleophilic attack, facilitating the formation of the C-C bond necessary for ring closure.
Following the initial coupling, the intermediate undergoes cyclization to form the isoxazole ring. The proposed mechanism suggests that the reaction passes through a difluoromethyl ketoxime species before final aromatization. This pathway avoids the formation of regioisomeric byproducts commonly seen in traditional hydroxylamine condensations, ensuring high purity of the final product. The mild reaction conditions prevent the decomposition of sensitive functional groups, allowing for the synthesis of complex molecules that would otherwise be inaccessible. Understanding this mechanism is vital for process chemists aiming to optimize reaction parameters for maximum yield and minimal impurity formation during scale-up.
How to Synthesize 3-Difluoromethylisoxazole Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires careful attention to reagent stoichiometry and reaction atmosphere. The standard protocol involves dissolving the terminal alkyne in chloroform, followed by the sequential addition of difluoroethylamine, tert-butyl nitrite, and acetic acid. The catalyst system, comprising cuprous iodide and zinc bromide, is then introduced under a nitrogen blanket to prevent oxidation of the copper species. The mixture is stirred at room temperature for approximately 24 hours, allowing the reaction to reach completion as monitored by TLC or HPLC.
- Dissolve the terminal alkyne substrate in chloroform and add difluoroethylamine, tert-butyl nitrite, and acetic acid under nitrogen protection.
- Introduce cuprous iodide and zinc bromide reagents to the mixture, ensuring full dissolution possibly with an ice-water bath initially.
- Stir the reaction at room temperature for 24 hours, then purify the crude product via flash column chromatography to obtain the target isoxazole.
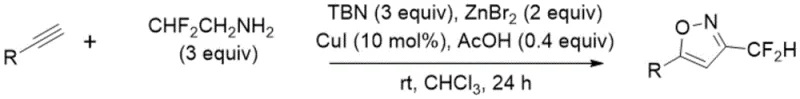
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain perspective, the adoption of this one-pot synthesis method offers transformative benefits that extend beyond mere chemical efficiency. The reliance on commercially available starting materials, such as difluoroethylamine and simple terminal alkynes, mitigates the risk of raw material shortages that often plague complex multi-step syntheses. This accessibility ensures a more resilient supply chain, capable of meeting the fluctuating demands of the pharmaceutical industry without significant lead time delays. Furthermore, the elimination of exotic or hazardous reagents simplifies logistics and storage requirements, reducing the overall overhead associated with chemical inventory management.
- Cost Reduction in Manufacturing: The economic impact of this technology is profound, primarily driven by the substitution of expensive precious metal catalysts with inexpensive copper and zinc salts. Traditional methods often require palladium or rhodium catalysts, which not only increase raw material costs but also necessitate rigorous metal scavenging steps to meet regulatory limits for residual metals in APIs. By utilizing base metal catalysis, this method inherently lowers the cost of goods sold (COGS) and simplifies the purification workflow. Additionally, the one-pot nature of the reaction reduces solvent usage and labor hours, contributing to substantial cost savings in large-scale production environments.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route translates directly into improved supply chain reliability. The mild reaction conditions and high functional group tolerance mean that the process is less susceptible to batch-to-batch variability caused by minor fluctuations in temperature or reagent quality. This consistency is critical for maintaining continuous manufacturing operations and meeting strict delivery schedules. Moreover, the high yields reported (ranging from 58% to 93%) ensure that less starting material is wasted, maximizing the output from each production run and securing a steady flow of high-purity intermediates for downstream drug synthesis.
- Scalability and Environmental Compliance: Scaling chemical processes from the bench to the plant floor often introduces unforeseen challenges, but this method is designed with scalability in mind. The absence of cryogenic conditions and hazardous oxidants like NCS makes the process safer and easier to manage in large reactors. From an environmental standpoint, the reduction in synthetic steps and waste generation aligns with green chemistry principles, helping manufacturers comply with increasingly stringent environmental regulations. This eco-friendly profile not only reduces waste disposal costs but also enhances the corporate sustainability image of the manufacturing entity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthesis technology. These insights are derived directly from the experimental data and comparative studies presented in the patent documentation, providing a clear understanding of the method's capabilities and limitations for potential adopters.
Q: What are the primary advantages of this one-pot method over traditional multi-step syntheses?
A: This method eliminates the need for pre-synthesizing complex intermediates like silyl enol ethers or difluoromethyl ketoximes. It utilizes commercially available difluoroethylamine and operates under mild room temperature conditions, significantly reducing operational complexity and waste generation compared to harsh cryogenic or multi-step oxidative protocols.
Q: Is this synthesis method suitable for large-scale commercial production?
A: Yes, the patent explicitly demonstrates gram-order synthesis capabilities with high yields ranging from 58% to 93%. The use of inexpensive copper and zinc catalysts, combined with simple workup procedures like reduced pressure distillation and flash chromatography, makes it highly amenable to commercial scale-up for API intermediate manufacturing.
Q: What is the scope of substrate tolerance for this reaction?
A: The reaction exhibits excellent functional group tolerance, successfully accommodating electron-withdrawing groups (nitro, cyano, trifluoromethyl), electron-donating groups (methoxy, alkyl), and heterocycles (thiophene, furan, pyridine). This broad scope allows for the rapid generation of diverse compound libraries for drug screening.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Difluoromethylisoxazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the success of drug development programs. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met with precision and reliability. We are committed to delivering products that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. By leveraging advanced technologies like the one described in CN108069918B, we can offer cost-effective solutions without compromising on the quality essential for pharmaceutical applications.
We invite you to collaborate with us to explore how this innovative synthesis route can benefit your specific pipeline. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements and timeline. Please contact us to request specific COA data and route feasibility assessments for your target molecules. Let us be your partner in navigating the complexities of fine chemical manufacturing, ensuring a seamless supply of critical building blocks for your next breakthrough therapy.