Advanced Synthesis of 2-Bromo-6-Fluoroanisole: Enhancing Safety and Scalability for Global Pharma Supply Chains
The pharmaceutical and fine chemical industries are constantly seeking robust synthetic pathways that balance high purity with operational safety, a challenge effectively addressed by the recent technological advancements disclosed in patent CN113372200B. This patent introduces a novel preparation method for 2-bromo-6-fluoroanisole, a critical building block in the synthesis of various bioactive molecules and agrochemical agents. Unlike traditional approaches that rely on hazardous nitration protocols and toxic alkylating agents, this innovative route utilizes 3,4-difluoronitrobenzene as a stable starting material, proceeding through a sequence of electrophilic substitution, nucleophilic displacement, reduction, and deamination. For R&D directors and procurement specialists, this shift represents a significant opportunity to optimize supply chain reliability while mitigating the environmental and safety risks associated with legacy manufacturing processes. The method not only ensures a high-purity profile suitable for sensitive downstream applications but also offers a scalable framework that aligns with modern green chemistry principles.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of fluorinated anisole derivatives has been plagued by severe safety and environmental drawbacks inherent to early-stage functionalization strategies. As illustrated in the prior art routes, conventional methods often commence with the direct nitration of fluorophenols using concentrated nitric acid, a process notorious for its exothermic volatility and potential for runaway reactions. Furthermore, subsequent methylation steps frequently employ methyl iodide, a reagent classified as highly toxic and carcinogenic, necessitating stringent containment measures and complex waste stream management. These factors collectively inflate the operational expenditure (OPEX) due to the need for specialized explosion-proof infrastructure and extensive effluent treatment, creating bottlenecks in the commercial scale-up of complex fluorinated intermediates. The reliance on such dangerous reagents not only jeopardizes worker safety but also introduces variability in product quality due to the formation of difficult-to-remove oxidative by-products.
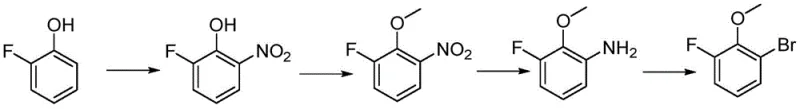
The Novel Approach
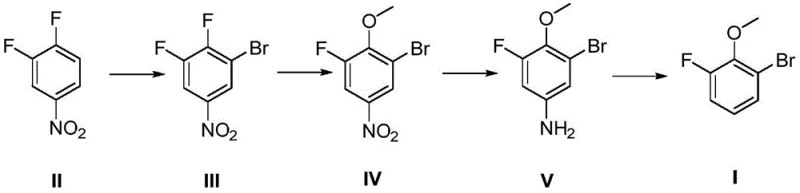
In stark contrast, the methodology outlined in CN113372200B circumvents these pitfalls by reversing the order of functional group introduction, starting with a commercially available nitro-substituted scaffold. By initiating the synthesis with an electrophilic bromination of 3,4-difluoronitrobenzene using N-bromosuccinimide (NBS), the process achieves high regioselectivity without the need for aggressive nitrating mixtures. This is followed by a mild nucleophilic aromatic substitution with sodium methoxide, which safely installs the methoxy moiety under controlled thermal conditions. The subsequent reduction of the nitro group and final deamination via diazotization proceed with high efficiency, yielding a product profile that is significantly cleaner than that obtained through phenol-based routes. This strategic reordering of synthetic steps effectively eliminates the use of explosive nitric acid and toxic methyl halides, thereby streamlining the regulatory approval process for high-purity 2-bromo-6-fluoroanisole intended for pharmaceutical use.
Mechanistic Insights into Electrophilic Substitution and Diazotization
The core of this synthetic innovation lies in the precise control of electronic effects during the initial bromination step. In the presence of concentrated sulfuric acid, the electron-withdrawing nitro group on the 3,4-difluoronitrobenzene ring directs the electrophilic attack of the bromonium ion (generated from NBS) to the position ortho to the nitro group and para to one of the fluorine atoms. This electrophilic aromatic substitution is highly favored due to the activation of the ring by the fluorine substituents despite the deactivating nature of the nitro group, ensuring that the bromine atom is installed at the correct position (Compound III) with minimal isomeric impurities. The reaction conditions, maintained between 20-60°C, allow for a kinetic control that prevents poly-bromination, a common side reaction in less optimized protocols. This selectivity is crucial for maintaining the integrity of the fluorine substituents, which are essential for the biological activity of the final API intermediates.
Following the bromination, the transformation of the nitro group to an amine and its subsequent removal via diazotization serves as a powerful strategy for structural editing. The reduction using sodium hydrosulfite is a clean, heterogeneous process that avoids the heavy metal contamination often associated with catalytic hydrogenation, thus simplifying the purification workflow. The final step involves the formation of a diazonium salt using isoamyl nitrite in DMF, which then undergoes a radical-mediated deamination to replace the amino group with a hydrogen atom. This deamination step is particularly elegant as it effectively removes the directing group (the nitro/amino functionality) that was necessary for the earlier regioselective bromination, leaving behind the desired 2-bromo-6-fluoroanisole skeleton. This mechanism ensures that the final product is free from nitrogenous impurities, meeting the stringent specifications required by global reliable pharma intermediate suppliers.
How to Synthesize 2-Bromo-6-Fluoroanisole Efficiently
The execution of this four-step sequence requires careful attention to stoichiometry and thermal management to maximize the overall yield, which the patent reports to be approximately 44% with individual step yields exceeding 79%. The process begins with the suspension of 3,4-difluoronitrobenzene in 98% sulfuric acid, followed by the controlled addition of NBS to manage the exotherm. Subsequent steps involve straightforward workups, such as quenching into ice water and recrystallization, which are easily adaptable to multi-kilogram scales. For detailed operational parameters including specific molar ratios, solvent choices, and reaction times that ensure reproducibility, please refer to the standardized protocol below.
- Perform electrophilic substitution on 3,4-difluoronitrobenzene using N-bromosuccinimide (NBS) in concentrated sulfuric acid to generate the brominated intermediate.
- React the brominated intermediate with sodium methoxide in methanol to introduce the methoxy group via nucleophilic aromatic substitution.
- Reduce the nitro group to an amine using sodium hydrosulfite, followed by diazotization and deamination with isoamyl nitrite to yield the final product.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic sourcing perspective, the adoption of this patented route offers compelling advantages that directly address the pain points of cost volatility and supply discontinuity in the fine chemical sector. By eliminating the dependency on hazardous reagents like methyl iodide and nitric acid, manufacturers can significantly reduce the overhead costs associated with safety compliance, hazardous waste disposal, and insurance premiums. This reduction in regulatory burden translates into a more stable pricing structure for buyers, as the production process is less susceptible to shutdowns caused by environmental inspections or safety incidents. Furthermore, the use of inexpensive and readily available starting materials like 3,4-difluoronitrobenzene ensures that the cost reduction in pharmaceutical intermediates manufacturing is sustainable over the long term, shielding customers from the price fluctuations often seen with specialty reagents.
- Cost Reduction in Manufacturing: The elimination of toxic methylating agents and explosive nitrating mixtures removes the need for expensive corrosion-resistant reactors and complex scrubbing systems, leading to substantial capital expenditure (CAPEX) savings. Additionally, the high selectivity of the NBS bromination minimizes the formation of by-products, reducing the solvent and energy consumption required for purification, which further drives down the unit cost of production.
- Enhanced Supply Chain Reliability: Because the raw materials employed in this route are commodity chemicals with robust global supply chains, the risk of production delays due to raw material shortages is drastically minimized. The simplified reaction sequence also shortens the overall cycle time, allowing suppliers to respond more agilely to demand spikes and ensuring consistent availability of high-purity 2-bromo-6-fluoroanisole for downstream drug synthesis.
- Scalability and Environmental Compliance: The process is inherently greener, generating significantly less acidic and toxic wastewater compared to traditional phenol nitration routes. This environmental compatibility facilitates easier permitting for capacity expansion, enabling manufacturers to scale from pilot batches to multi-ton production without encountering the regulatory hurdles that typically stall the commercial scale-up of complex fluorinated intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis method, providing clarity on its feasibility and benefits for industrial partners. These insights are derived directly from the experimental data and comparative analysis presented in the patent documentation, ensuring that decision-makers have accurate information for their evaluation.
Q: Why is the new synthesis route for 2-bromo-6-fluoroanisole considered safer than conventional methods?
A: The novel route avoids the use of concentrated nitric acid for direct nitration, which poses significant explosion risks, and eliminates the need for highly toxic methyl iodide in the methylation step, thereby enhancing operational safety and reducing hazardous waste generation.
Q: What are the typical yield characteristics of this patented preparation method?
A: According to the patent data, the process demonstrates robust efficiency with the lowest yield in any single step reaching approximately 79%, resulting in a respectable total yield of about 44% across the four-step sequence.
Q: How does this method impact the cost structure for pharmaceutical intermediate manufacturing?
A: By utilizing low-cost starting materials like 3,4-difluoronitrobenzene and avoiding expensive purification steps associated with toxic reagents, the method significantly lowers raw material and waste treatment costs, offering a more economically viable pathway for large-scale production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Bromo-6-Fluoroanisole Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition to safer and more efficient synthetic routes is critical for maintaining competitiveness in the global pharmaceutical market. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of patents like CN113372200B are fully realized in practical, large-volume manufacturing. We operate stringent purity specifications and maintain rigorous QC labs to guarantee that every batch of 2-bromo-6-fluoroanisole meets the exacting standards required for API synthesis, providing our partners with the confidence needed to accelerate their own drug development timelines.
We invite procurement leaders and technical directors to engage with us for a Customized Cost-Saving Analysis tailored to your specific volume requirements. By leveraging our optimized production capabilities, you can secure a stable supply of this key intermediate while achieving significant economic efficiencies. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how our commitment to innovation can drive value for your organization.