Advanced One-Step Synthesis of 5-Chloro-2-(pyridin-3-yl)pyridin-3-amine for Commercial Scale-Up
The pharmaceutical industry constantly seeks efficient pathways to construct complex heterocyclic scaffolds, particularly biaryl amines which serve as critical building blocks for kinase inhibitors and other therapeutic agents. Patent CN111303020A introduces a groundbreaking synthetic methodology for the preparation of 5-chloro-2-(pyridin-3-yl)pyridin-3-amine, a valuable intermediate in modern drug discovery. This innovation addresses the longstanding challenges associated with constructing sterically hindered pyridine-pyridine bonds by leveraging a highly optimized palladium-catalyzed cross-coupling strategy. Unlike traditional multi-step sequences that suffer from cumulative yield losses and extensive waste generation, this novel approach streamlines the synthesis into a single, robust operational step. By utilizing readily accessible halo-pyridine precursors and boronic acids, the method ensures high atom economy and operational simplicity, positioning it as a superior choice for reliable pharmaceutical intermediate supplier networks aiming to enhance their portfolio efficiency.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of substituted biaryl amines like 5-chloro-2-(pyridin-3-yl)pyridin-3-amine has been plagued by significant technical hurdles that impede cost-effective manufacturing. Conventional routes often rely on harsh nucleophilic aromatic substitution conditions that require extreme temperatures and pressures, leading to poor selectivity and the formation of difficult-to-remove regioisomers. Furthermore, many legacy processes necessitate the use of protecting groups to mask reactive amine functionalities during the coupling phase, adding at least two additional synthetic steps involving protection and subsequent deprotection. These extra operations not only drastically increase the consumption of reagents and solvents but also extend the overall production cycle time, creating bottlenecks in the supply chain. Additionally, the use of less active catalysts in older methodologies often results in incomplete conversions, requiring rigorous and expensive purification protocols such as preparative HPLC to meet the stringent purity specifications demanded by regulatory bodies for clinical applications.
The Novel Approach
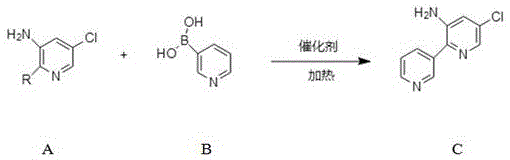
The methodology disclosed in the patent represents a paradigm shift by employing a direct Suzuki-Miyaura cross-coupling reaction that bypasses the need for protecting groups entirely. This novel approach utilizes a specific combination of a palladium chloride catalyst and a carbonate base in a dioxane-water solvent system to facilitate the bond formation under relatively mild thermal conditions. The strategic selection of 2-halo-5-chloropyridin-3-amine as the electrophile allows for precise control over the regioselectivity of the coupling, ensuring that the new carbon-carbon bond forms exclusively at the desired position. By optimizing the stoichiometry and reaction parameters, this route achieves exceptional conversion rates, with experimental examples demonstrating isolated yields approaching 95%. This simplification of the synthetic route not only reduces the physical footprint required for manufacturing but also significantly lowers the environmental impact by minimizing solvent usage and waste generation, aligning perfectly with green chemistry principles essential for sustainable cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Pd-Catalyzed Suzuki-Miyaura Coupling
The core of this synthetic breakthrough lies in the intricate catalytic cycle driven by the palladium species, which facilitates the cross-coupling between the aryl halide and the organoboron reagent. The mechanism initiates with the oxidative addition of the palladium(0) species into the carbon-halogen bond of the 2-bromo-5-chloropyridin-3-amine substrate, forming a reactive organopalladium(II) intermediate. This step is critical and is influenced heavily by the electronic nature of the pyridine ring; the presence of the electron-withdrawing chlorine atom at the 5-position enhances the electrophilicity of the carbon-bromine bond, thereby accelerating the oxidative addition phase. Following this, the base, specifically cesium carbonate in the optimized protocol, activates the pyridin-3-ylboronic acid by forming a more nucleophilic boronate species. This activated boronate then undergoes transmetallation with the palladium complex, transferring the pyridyl group to the metal center and creating a diaryl-palladium species. The cycle concludes with reductive elimination, where the two aryl groups couple to form the final biaryl product while regenerating the active palladium(0) catalyst to enter the next turnover. Understanding this cycle is vital for R&D teams to troubleshoot potential issues such as catalyst deactivation or homocoupling side reactions.
Impurity control is another critical aspect where this mechanistic understanding translates into practical quality assurance. A common challenge in Suzuki couplings is the protodeboronation of the boronic acid or the homocoupling of the aryl halide, both of which can generate structurally similar impurities that are difficult to separate. The patent specifies the use of a biphasic solvent system comprising dioxane and water, which plays a dual role in suppressing these side reactions. The aqueous phase ensures the solubility of the inorganic base, facilitating efficient activation of the boronic acid, while the organic phase solubilizes the hydrophobic reactants and the catalyst. Furthermore, the strict control of the nitrogen atmosphere prevents the oxidation of the phosphine ligands or the palladium center, which could otherwise lead to the precipitation of palladium black and cessation of the catalytic activity. By maintaining the reaction temperature within the narrow window of 80-100°C, the process balances the kinetic energy required for the coupling against the thermal degradation of sensitive intermediates, ensuring a clean reaction profile that simplifies downstream processing and guarantees high-purity pharmaceutical intermediates.
How to Synthesize 5-Chloro-2-(pyridin-3-yl)pyridin-3-amine Efficiently
Implementing this synthesis requires careful attention to the order of addition and the quality of reagents to maximize the yield and reproducibility of the process. The protocol begins with the precise weighing of the halo-pyridine substrate and the boronic acid coupling partner, followed by their dissolution in anhydrous dioxane to create a homogeneous reaction mixture. It is imperative to maintain an inert atmosphere throughout the procedure to protect the catalyst from oxidative degradation, which is achieved by purging the reactor with nitrogen prior to the addition of the aqueous base and catalyst solution. The reaction is then heated to the target temperature and monitored closely, typically reaching completion within a few hours, after which a straightforward workup involving aqueous quenching and organic extraction isolates the crude product. For detailed operational parameters and specific stoichiometric ratios validated at the 500g scale, please refer to the standardized synthesis guide below.
- Charge compound A (2-bromo-5-chloropyridin-3-amine) and compound B (pyridin-3-ylboronic acid) into a reactor with dioxane solvent under nitrogen protection.
- Add an aqueous solution of cesium carbonate and palladium chloride catalyst, then heat the mixture to 80-100°C for 2-4 hours with stirring.
- Upon completion, quench with water, extract with ethyl acetate, dry the organic layer, and purify via column chromatography to isolate the target amine.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the adoption of this synthetic route offers transformative benefits that directly impact the bottom line and operational resilience. The elimination of multiple synthetic steps inherently reduces the consumption of raw materials, solvents, and energy, leading to substantial cost savings in the overall manufacturing budget without compromising on quality. Moreover, the reliance on commodity chemicals such as cesium carbonate, palladium chloride, and dioxane ensures that the supply chain is not vulnerable to the volatility associated with exotic or proprietary reagents. This stability allows procurement managers to secure long-term contracts with reliable suppliers, mitigating the risk of production stoppages due to material shortages. The robustness of the reaction conditions also implies that the process can be transferred between different manufacturing sites with minimal re-validation, enhancing the flexibility of the global supply network.
- Cost Reduction in Manufacturing: The streamlined one-step nature of this process eliminates the need for intermediate isolation and purification stages that are typical in multi-step syntheses. By removing these unit operations, manufacturers save significantly on labor costs, equipment occupancy time, and solvent recovery expenses. The high yield reported in the patent examples means that less starting material is required to produce the same amount of final product, effectively lowering the cost of goods sold (COGS). Furthermore, the simplified workup procedure reduces the load on waste treatment facilities, contributing to lower environmental compliance costs and avoiding the financial penalties associated with hazardous waste disposal.
- Enhanced Supply Chain Reliability: The use of 2-bromo-5-chloropyridin-3-amine and pyridin-3-ylboronic acid as starting materials leverages a mature global market for pyridine derivatives. These precursors are produced by multiple vendors worldwide, reducing the risk of single-source dependency. The moderate reaction conditions (80-100°C) do not require specialized high-pressure or cryogenic equipment, meaning that the synthesis can be performed in standard glass-lined or stainless steel reactors available in most contract manufacturing organizations. This compatibility with existing infrastructure accelerates the timeline from process development to commercial production, ensuring that project milestones are met consistently and reducing lead time for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: The patent data demonstrates successful execution at the 500g scale, providing a solid foundation for scaling up to multi-kilogram or ton-level production. The biphasic solvent system allows for efficient heat transfer and mixing, which are critical factors when moving from laboratory to pilot and commercial scales. Additionally, the process generates fewer byproducts compared to traditional methods, simplifying the effluent profile and making it easier to meet increasingly stringent environmental regulations. The ability to recycle the organic solvent (dioxane) further enhances the sustainability profile of the process, appealing to partners who prioritize green chemistry initiatives in their vendor selection criteria.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and technical disclosures within the patent documentation to provide clarity for potential partners. Understanding these details is crucial for evaluating the feasibility of integrating this route into your existing manufacturing capabilities.
Q: What is the primary advantage of this synthetic route over conventional methods?
A: The primary advantage is the reduction to a single-step reaction with a short route, eliminating the need for complex multi-step protection and deprotection sequences often required in traditional biaryl amine synthesis.
Q: Which halogen substituent on the pyridine ring provides the optimal yield?
A: Experimental data indicates that using the bromo-substituted precursor (2-bromo-5-chloropyridin-3-amine) combined with cesium carbonate yields the highest conversion rates, achieving up to 94.98% isolated yield.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process utilizes easily available raw materials, standard solvents like dioxane, and operates at moderate temperatures (80-100°C), making it highly adaptable for commercial scale-up with easy reaction control.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Chloro-2-(pyridin-3-yl)pyridin-3-amine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that efficient synthetic routes play in accelerating drug development timelines. Our team of expert chemists has thoroughly analyzed the patented methodology for 5-chloro-2-(pyridin-3-yl)pyridin-3-amine and is fully prepared to execute this process with the highest standards of quality and safety. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met whether you are in the pre-clinical phase or preparing for market launch. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of material we deliver meets the exacting requirements of the global pharmaceutical industry.
We invite you to collaborate with us to optimize your supply chain for this key intermediate. By leveraging our technical expertise, we can provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us help you secure a stable, high-quality supply of 5-chloro-2-(pyridin-3-yl)pyridin-3-amine, allowing you to focus on your core mission of developing life-saving therapies.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →