Advanced Synthesis of Relugolix Impurities for Enhanced Quality Control and Commercial Scalability
Advanced Synthesis of Relugolix Impurities for Enhanced Quality Control and Commercial Scalability
The pharmaceutical landscape for gonadotropin-releasing hormone (GnRH) receptor antagonists has been significantly advanced by the introduction of Relugolix, a potent therapeutic agent developed for treating uterine fibroids and prostate cancer. As depicted in the molecular structure below, the complexity of this molecule necessitates rigorous quality control measures to ensure patient safety and efficacy. However, the synthesis of such complex heterocyclic systems often generates trace impurities that are structurally similar to the active pharmaceutical ingredient (API), posing significant challenges for regulatory compliance and batch release. Patent CN114805326A addresses a critical gap in this domain by disclosing a robust preparation process for Relugolix Impurity I and Impurity II, which are known to form during standard manufacturing but are notoriously difficult to isolate and characterize.
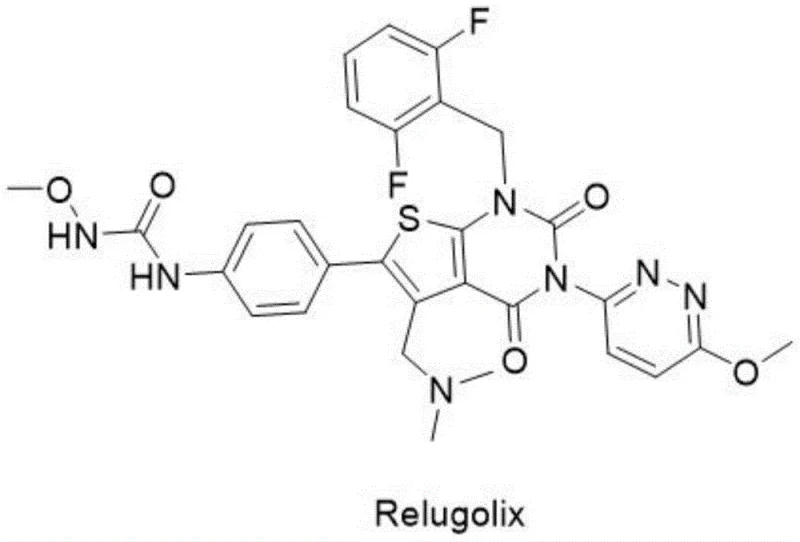
This patent represents a pivotal development for analytical chemists and quality assurance teams, providing a definitive synthetic route to generate these specific impurities as high-purity reference standards. By establishing a reliable method to produce these compounds, manufacturers can implement more accurate high-performance liquid chromatography (HPLC) methods for quantifying impurities in the final bulk drug. This capability is essential for any reliable pharmaceutical intermediate supplier aiming to support global regulatory filings, as the ability to detect and control genotoxic or persistent impurities is a cornerstone of modern Good Manufacturing Practice (GMP). The disclosed technology not only solves the problem of reference material scarcity but also offers insights into the degradation pathways of the drug substance itself.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
In the conventional manufacturing of Relugolix, the formation of side products is an inherent risk due to the multifunctional nature of the intermediates involved. Historically, the industry has relied on isolating these impurities from crude reaction mixtures, a process that is fraught with inefficiency and inconsistency. As illustrated in the traditional synthetic pathway, partial hydrolysis or unintended condensation reactions can occur under standard processing conditions, leading to the formation of Impurity II and subsequently Impurity I. These byproducts are often present in very low concentrations, making their isolation via preparative HPLC or crystallization economically unviable and technically demanding. Furthermore, without authentic standards, identifying these peaks in a chromatogram relies on speculative structural elucidation, which introduces uncertainty into the quality control process.
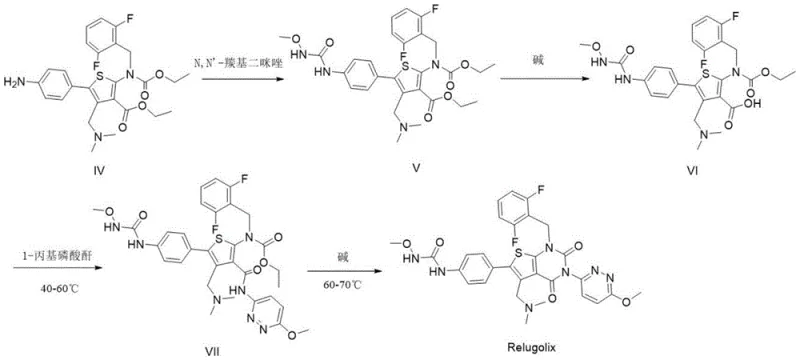
The reliance on isolation means that supply chains for these critical reference standards are fragile; if a production batch of the API does not generate enough of the specific impurity, the QC lab cannot calibrate their instruments. This bottleneck can delay batch releases and complicate stability studies required for regulatory submissions. Additionally, the structural similarity of these impurities to the parent compound makes separation difficult, often requiring specialized chiral columns or complex mobile phases that increase the cost of analysis. For procurement managers, this translates to higher operational expenditures and increased risk of batch rejection if impurity levels drift beyond specification limits without a clear understanding of their identity.
The Novel Approach
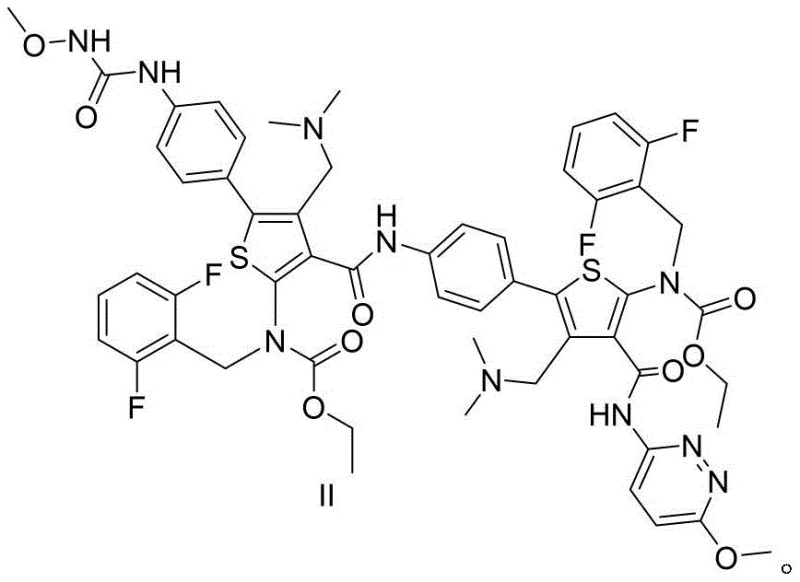
The patented methodology introduces a directed, step-wise synthesis that bypasses the inefficiencies of isolation by constructing the impurity molecules from specific precursors. As shown in the reaction scheme below, the process begins with a controlled condensation between Compound VI and Compound III, utilizing 1-propyl phosphoric anhydride (T3P) as a coupling agent. This reaction is conducted under mild conditions, typically between 20-30°C, in solvents such as ethyl acetate or tetrahydrofuran, ensuring high selectivity for Impurity II. This targeted approach eliminates the variability associated with harvesting byproducts from random side reactions, guaranteeing a consistent supply of the intermediate.
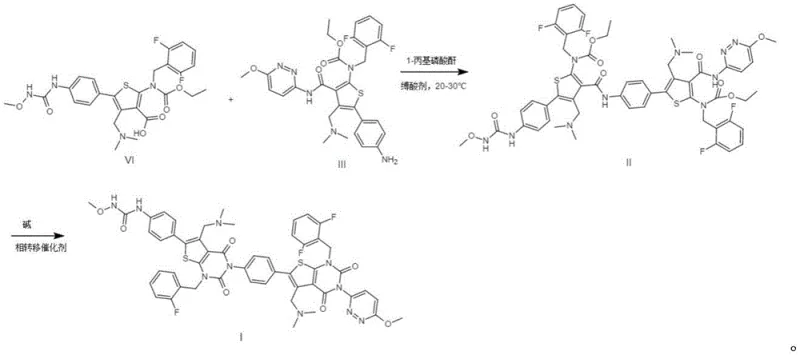
Following the formation of Impurity II, the process employs a subsequent cyclization step to generate Impurity I. This transformation is catalyzed by a base and a phase transfer catalyst at elevated temperatures of 45-55°C, driving the reaction to completion with impressive yields exceeding 80%. This two-step sequence is a paradigm shift from passive isolation to active synthesis, empowering manufacturers to produce gram-to-kilogram quantities of these reference standards on demand. For a reliable pharmaceutical intermediate supplier, this route offers a scalable solution that decouples the supply of QC standards from the fluctuations of API production batches, thereby stabilizing the entire quality assurance workflow.
Mechanistic Insights into T3P-Mediated Condensation and Cyclization
The core of this innovative synthesis lies in the strategic use of 1-propyl phosphoric anhydride (T3P) to facilitate the amide bond formation between the carboxylic acid moiety of Compound VI and the amine group of Compound III. T3P is a superior coupling reagent known for its ability to activate carboxylic acids while minimizing racemization and side reactions, which is critical when dealing with complex chiral molecules like Relugolix derivatives. The reaction mechanism involves the formation of a mixed anhydride intermediate, which is then attacked by the nucleophilic amine to form the desired peptide-like bond found in Impurity II. The choice of solvent, such as acetonitrile or ethyl acetate, plays a vital role in solubilizing the reactants and managing the exotherm, ensuring that the reaction proceeds smoothly without degrading the sensitive fluorinated aromatic rings present in the structure.
Once Impurity II is secured, the conversion to Impurity I involves an intramolecular cyclization that closes the ring system characteristic of the final impurity structure. This step requires precise control of basicity and temperature to promote nucleophilic attack without causing hydrolysis of the newly formed amide bond. The addition of phase transfer catalysts like tetrabutylammonium bromide enhances the reactivity of the base in organic media, facilitating the deprotonation of the nucleophile and accelerating the ring closure. Understanding this mechanistic pathway is crucial for R&D directors, as it highlights the specific conditions under which these impurities form in the main API process, allowing for proactive mitigation strategies such as adjusting pH or temperature profiles during the main synthesis to suppress their generation.
How to Synthesize Relugolix Impurities Efficiently
The execution of this synthesis requires strict adherence to the optimized parameters defined in the patent to achieve the reported high purity and yield. The process is designed to be operationally simple, utilizing commercially available reagents and standard laboratory equipment, which facilitates easy technology transfer from R&D to pilot plant scales. Operators must maintain an inert nitrogen atmosphere during the coupling step to prevent moisture interference, which could deactivate the T3P reagent. Following the reaction, a straightforward workup involving aqueous quenching and organic extraction allows for the removal of water-soluble byproducts, while final purification via column chromatography ensures the reference standard meets the stringent purity requirements necessary for analytical validation.
- Condense Compound VI and Compound III using 1-propyl phosphoric anhydride (T3P) in solvents like ethyl acetate or THF at 20-30°C to obtain Impurity II.
- Purify the resulting Impurity II via extraction and column chromatography to achieve purity levels exceeding 96%.
- Subject Impurity II to cyclization using a base and phase transfer catalyst at 45-55°C to generate Impurity I with high yield.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the implementation of this patented synthesis route offers substantial strategic benefits beyond mere technical feasibility. The ability to internally produce or source high-purity impurity standards significantly de-risks the supply chain for the final API. When a manufacturer can guarantee the availability of reference standards, they reduce the lead time for method validation and stability testing, which are often critical path items in drug development timelines. This reliability translates directly into faster time-to-market for generic versions or new formulations of Relugolix, providing a competitive edge in the marketplace. Furthermore, the high yields reported in the patent suggest a cost-effective production model, reducing the unit cost of these high-value reference materials.
- Cost Reduction in Manufacturing: The elimination of complex isolation procedures and the use of efficient coupling reagents like T3P drastically simplify the production workflow. By avoiding the need for extensive preparative HPLC purification which consumes large volumes of solvents and stationary phases, the overall cost of goods sold (COGS) for these reference standards is significantly lowered. This efficiency allows for the allocation of resources towards other critical quality initiatives, optimizing the overall operational budget without compromising on the integrity of the analytical data.
- Enhanced Supply Chain Reliability: Dependence on isolating impurities from API batches creates a single point of failure in the supply chain; if the API process is optimized to reduce impurities, the reference standard supply dries up. This synthetic route breaks that dependency, ensuring a continuous and predictable supply of Impurity I and II regardless of API batch performance. This stability is vital for maintaining uninterrupted QC operations and ensuring that regulatory audits can be supported with valid, traceable reference materials at any time.
- Scalability and Environmental Compliance: The reaction conditions described, operating at moderate temperatures and using common organic solvents, are inherently scalable from gram to multi-kilogram batches. This scalability supports the growing demand for Relugolix as its indications expand globally. Moreover, the high atom economy of the coupling reaction and the ability to recycle solvents contribute to a greener manufacturing profile, aligning with modern environmental, social, and governance (ESG) goals that are increasingly important for corporate procurement policies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these specific Relugolix impurities. Understanding the nuances of this synthesis helps stakeholders make informed decisions about their quality control strategies and supplier partnerships. The answers provided are derived directly from the technical specifications and experimental data outlined in the patent documentation, ensuring accuracy and relevance for industry professionals.
Q: Why are Relugolix Impurity I and II critical for quality control?
A: These impurities are difficult to remove during standard synthesis and can persist in the bulk drug. Having authentic reference standards allows for accurate quantitative detection, ensuring the final API meets stringent safety regulations.
Q: What is the advantage of the patented synthesis route over isolation?
A: Isolating these impurities from reaction mixtures is inefficient and yields low quantities. The patented directed synthesis provides high yields (over 80%) and high purity (>95%), ensuring a reliable supply of reference materials.
Q: Can this process be scaled for commercial reference standard production?
A: Yes, the process utilizes common reagents like T3P and standard bases under mild temperatures (20-55°C), making it highly amenable to scale-up from gram to kilogram scales for industrial QC needs.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Relugolix Impurity Supplier
At NINGBO INNO PHARMCHEM, we recognize that the quality of an API is only as good as the standards used to measure it. Our team of expert chemists has extensively analyzed the pathway described in CN114805326A and possesses the technical capability to execute this synthesis with precision. We offer comprehensive support for the scale-up of these complex intermediates, leveraging our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying the structural integrity and purity of every batch, ensuring that our clients receive materials that meet the most stringent purity specifications required for global regulatory submissions.
We invite pharmaceutical companies and CROs to collaborate with us to optimize their supply chain for Relugolix impurities. By partnering with us, you gain access to a Customized Cost-Saving Analysis that evaluates how integrating our synthesized standards can streamline your QC workflows. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments tailored to your project needs. Let us help you secure your supply chain and accelerate your path to market with confidence.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →