Revolutionizing Isoindoline-1-Carboxylic Acid Production via One-Step Cyclization for Commercial Scale
Introduction to Advanced Isoindoline Synthesis Technology
The landscape of pharmaceutical intermediate manufacturing is constantly evolving, driven by the need for more efficient, cost-effective, and scalable synthetic routes. A pivotal advancement in this domain is detailed in patent CN111718290B, which discloses a novel synthesis method for multi-configuration isoindoline-1-carboxylic acid amino acid compounds. This technology represents a paradigm shift from traditional multi-step linear syntheses to a highly convergent, one-step cyclization strategy. For R&D directors and process chemists, this development offers a compelling solution to the longstanding challenges associated with constructing the isoindoline core, a critical scaffold found in numerous bioactive molecules including hepatitis C virus inhibitors. The ability to access these valuable non-natural amino acids through a streamlined process not only accelerates drug discovery timelines but also lays a robust foundation for commercial manufacturing.
The significance of this patent lies in its direct address of inefficiencies inherent in prior art. By utilizing readily available phenylglycine derivatives and formaldehyde under acidic conditions, the method bypasses the need for complex protecting group strategies and harsh cryogenic operations. This approach aligns perfectly with the principles of green chemistry and process intensification, making it an attractive candidate for industrial adoption. As a reliable pharmaceutical intermediate supplier, understanding and leveraging such technological breakthroughs is essential for maintaining competitiveness in the global market. The following analysis delves deep into the mechanistic nuances, comparative advantages, and commercial implications of this innovative synthetic pathway.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the innovations described in CN111718290B, the synthesis of isoindoline-1-carboxylic acid derivatives was fraught with operational complexities and economic inefficiencies. As illustrated in the background art referenced by the patent, conventional routes often relied on starting materials like 5-bromoisoindoline-1-one, necessitating a tedious five-step reaction sequence to reach the final carboxylic acid product. 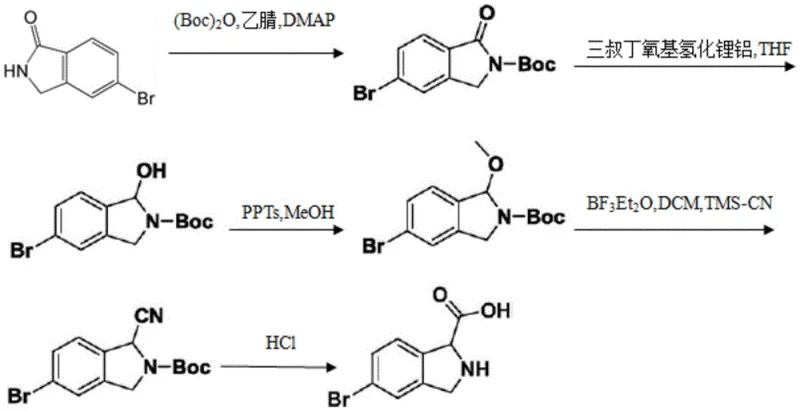 This legacy approach suffered from several critical drawbacks that hindered scalability. Firstly, the requirement for cryogenic conditions, specifically stirring operations at -78°C in certain steps, imposed a massive burden on energy consumption and specialized equipment requirements. Secondly, the cumulative yield of a five-step process is inherently low; even with moderate yields per step, the overall throughput diminishes significantly, leading to excessive waste generation and higher cost of goods sold (COGS). Furthermore, the use of multiple solvents and reagents across different stages complicated the purification workflow, often requiring extensive chromatographic separations that are difficult to translate to ton-scale production.
This legacy approach suffered from several critical drawbacks that hindered scalability. Firstly, the requirement for cryogenic conditions, specifically stirring operations at -78°C in certain steps, imposed a massive burden on energy consumption and specialized equipment requirements. Secondly, the cumulative yield of a five-step process is inherently low; even with moderate yields per step, the overall throughput diminishes significantly, leading to excessive waste generation and higher cost of goods sold (COGS). Furthermore, the use of multiple solvents and reagents across different stages complicated the purification workflow, often requiring extensive chromatographic separations that are difficult to translate to ton-scale production.
The Novel Approach
In stark contrast to the cumbersome legacy methods, the novel approach disclosed in the patent simplifies the entire synthetic logic into a single, elegant transformation. The core innovation involves the direct reaction of a compound of Formula I (a substituted phenylglycine) with formaldehyde under acidic conditions to yield the target isoindoline-1-carboxylic acid (Formula II). 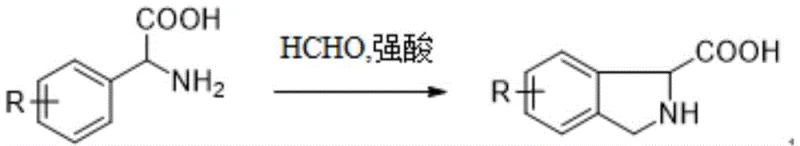 This biomimetic cyclization, reminiscent of a Pictet-Spengler reaction, proceeds with remarkable efficiency. The reaction conditions are mild, typically requiring heating to only 80-90°C followed by stirring at ambient temperature, completely eliminating the need for cryogenic cooling. The simplicity of this one-pot procedure drastically reduces the number of unit operations, solvent swaps, and intermediate isolations. From a process chemistry perspective, this convergence translates to a substantial reduction in manufacturing lead time and a significant improvement in overall space-time yield. The robustness of this method allows for the direct conversion of diverse substrates, including those with electron-withdrawing and electron-donating groups, without compromising the integrity of the sensitive amino acid functionality.
This biomimetic cyclization, reminiscent of a Pictet-Spengler reaction, proceeds with remarkable efficiency. The reaction conditions are mild, typically requiring heating to only 80-90°C followed by stirring at ambient temperature, completely eliminating the need for cryogenic cooling. The simplicity of this one-pot procedure drastically reduces the number of unit operations, solvent swaps, and intermediate isolations. From a process chemistry perspective, this convergence translates to a substantial reduction in manufacturing lead time and a significant improvement in overall space-time yield. The robustness of this method allows for the direct conversion of diverse substrates, including those with electron-withdrawing and electron-donating groups, without compromising the integrity of the sensitive amino acid functionality.
Mechanistic Insights into Acid-Catalyzed Cyclization
The chemical elegance of this synthesis lies in its mechanism, which leverages the nucleophilicity of the aromatic ring and the electrophilicity of the iminium ion generated in situ. Under strong acidic conditions, such as those provided by 4M hydrochloric acid or hydrobromic acid, the primary amine of the phenylglycine derivative reacts rapidly with formaldehyde to form an iminium intermediate. This highly electrophilic species is then attacked by the electron-rich aromatic ring at the ortho-position, facilitating ring closure to form the isoindoline skeleton. The subsequent hydrolysis or stabilization steps yield the final carboxylic acid product. This mechanism is highly tolerant of various substituents on the benzene ring, as evidenced by the successful synthesis of derivatives bearing methyl, fluoro, chloro, and bromo groups. The preservation of stereochemistry is another critical mechanistic feature; when chiral starting materials like (R)-2-amino-2-phenylacetic acid are employed, the cyclization proceeds with retention of configuration, yielding enantiomerically pure (R)-isoindoline-1-carboxylic acid. 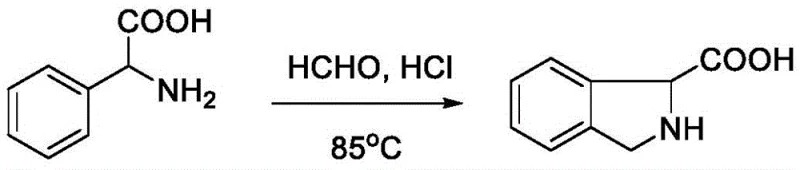 This stereospecificity is paramount for pharmaceutical applications where the biological activity is often dependent on a specific enantiomer.
This stereospecificity is paramount for pharmaceutical applications where the biological activity is often dependent on a specific enantiomer.
Impurity control is inherently superior in this one-step design compared to multi-step alternatives. In linear syntheses, impurities generated in early steps can propagate and amplify through subsequent reactions, creating complex impurity profiles that are difficult to purge. By condensing the synthesis into a single vessel, the potential for side reactions is minimized, and the impurity profile is significantly cleaner. The use of aqueous acid media also facilitates easy workup procedures; post-reaction, the pH can be adjusted to precipitate the product or facilitate extraction, avoiding the need for complex organic workups often associated with organometallic or anhydrous conditions. The high yields reported, consistently exceeding 82 percent across various substrates, indicate that the reaction equilibrium strongly favors the cyclized product, further simplifying the purification burden and ensuring high purity specifications suitable for GMP manufacturing environments.
How to Synthesize Isoindoline-1-Carboxylic Acid Efficiently
Implementing this synthesis route in a laboratory or pilot plant setting requires careful attention to reaction parameters to maximize yield and safety. The process begins with the dissolution of the phenylglycine starting material in a concentrated strong acid, followed by the controlled addition of formaldehyde. The reaction is exothermic initially and requires thermal management to maintain the optimal temperature window of 80-90°C. Following the initial heating phase, a prolonged stirring period at room temperature ensures complete conversion. The detailed standardized operating procedures, including specific quenching protocols and crystallization conditions for different derivatives, are critical for reproducibility. For a comprehensive guide on executing this synthesis with precision, please refer to the step-by-step instructions provided below.
- Dissolve the starting phenylglycine derivative (Formula I) in a concentrated strong acid solution such as 4M hydrochloric acid or hydrobromic acid.
- Add an aqueous formaldehyde solution (30-45% concentration) to the reaction mixture with a molar ratio of formaldehyde to substrate ranging from 3: 1 to 5:1.
- Heat the mixture to 80-90°C for 3-5 hours, then cool to room temperature (15-35°C) and stir for an additional 10-14 hours to complete the cyclization.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this novel synthesis methodology offers transformative benefits that extend far beyond simple chemical efficiency. The primary value driver is the drastic simplification of the supply chain. By replacing a five-step sequence with a single reaction, the number of raw materials required is significantly reduced. This consolidation minimizes the risk of supply disruptions associated with sourcing multiple specialized intermediates and solvents. Furthermore, the starting materials—substituted phenylglycines and formaldehyde—are commodity chemicals available from a wide range of global suppliers, ensuring robust supply continuity and competitive pricing. This shift effectively decouples production capacity from the bottlenecks of complex multi-step synthesis, allowing for more flexible and responsive manufacturing schedules.
- Cost Reduction in Manufacturing: The economic impact of this process innovation is profound. Eliminating four reaction steps removes the associated costs of labor, energy, solvent consumption, and waste disposal for those stages. Specifically, the removal of cryogenic steps (-78°C) eliminates the need for expensive cooling baths and specialized low-temperature reactors, resulting in substantial capital expenditure (CAPEX) and operational expenditure (OPEX) savings. The high yield of the single-step reaction means less raw material is wasted per kilogram of product, directly lowering the variable cost of production. Additionally, the simplified purification process reduces the consumption of chromatography media and solvents, further driving down the cost of goods. These efficiencies collectively enable a much more competitive pricing structure for the final isoindoline intermediates.
- Enhanced Supply Chain Reliability: Reliability is the cornerstone of a resilient supply chain. The robustness of this one-pot synthesis makes it less susceptible to variability and failure modes common in long linear sequences. With fewer unit operations, there are fewer opportunities for human error or equipment malfunction. The use of stable, commercially available starting materials mitigates the risk of raw material shortages. Moreover, the shorter cycle time—from raw material intake to finished product—allows for faster turnaround on customer orders. This agility is crucial in the fast-paced pharmaceutical industry, where speed to market can determine the success of a drug development program. Manufacturers adopting this route can offer more reliable lead times and greater flexibility to accommodate fluctuating demand.
- Scalability and Environmental Compliance: Scaling chemical processes from the bench to the plant is often where traditional routes fail due to heat transfer limitations or safety concerns. This novel method, operating at mild temperatures and atmospheric pressure, scales up linearly with minimal engineering challenges. The reduction in solvent usage and waste generation aligns with increasingly stringent environmental regulations and corporate sustainability goals. By minimizing the E-factor (mass of waste per mass of product), manufacturers can reduce their environmental footprint and lower waste treatment costs. This eco-friendly profile not only ensures regulatory compliance but also enhances the brand reputation of the supplier as a responsible partner in sustainable chemistry.
Frequently Asked Questions (FAQ)
Understanding the technical and commercial nuances of this synthesis method is vital for stakeholders evaluating its adoption. The following questions address common inquiries regarding the scope, stereochemistry, and practical implementation of the technology described in patent CN111718290B. These answers are derived directly from the experimental data and claims within the patent, providing a factual basis for decision-making. Whether you are assessing the feasibility for a specific analog or evaluating the quality standards, this information serves as a foundational resource.
Q: What are the primary advantages of this new synthesis method over traditional routes?
A: The new method reduces a complex five-step sequence involving cryogenic conditions into a single pot reaction, significantly lowering energy consumption and operational complexity while improving overall yield to over 82 percent.
Q: Can this process accommodate chiral starting materials to produce enantiopure products?
A: Yes, the process preserves the stereochemistry of the starting material. Using (R)- or (S)-2-amino-2-phenylacetic acid derivatives directly yields the corresponding (R)- or (S)-isoindoline-1-carboxylic acid with high optical purity.
Q: What types of substituents are tolerated on the aromatic ring during this cyclization?
A: The reaction is highly robust and tolerates various substituents including hydrogen, methyl, methoxy, fluorine, chlorine, bromine, and trifluoromethyl groups at the 4, 5, 6, or 7 positions of the isoindoline ring.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Isoindoline-1-Carboxylic Acid Supplier
The technological advancements detailed in patent CN111718290B underscore the immense potential of isoindoline-1-carboxylic acid derivatives as key building blocks in modern medicinal chemistry. At NINGBO INNO PHARMCHEM, we recognize the critical importance of translating such innovative laboratory methods into robust, commercial-scale realities. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the specific requirements of this acid-catalyzed cyclization, ensuring that every batch meets stringent purity specifications. Our rigorous QC labs employ advanced analytical techniques to verify identity, potency, and chiral purity, guaranteeing that the intermediates we supply are perfectly suited for the synthesis of high-value APIs like hepatitis C inhibitors.
We invite pharmaceutical companies and research institutions to collaborate with us to leverage this efficient synthesis route for their projects. By partnering with NINGBO INNO PHARMCHEM, you gain access not just to a chemical product, but to a comprehensive service model focused on optimization and reliability. We encourage you to contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. Let us provide you with specific COA data and route feasibility assessments to demonstrate how we can accelerate your development timeline while optimizing your budget. Together, we can drive the next generation of therapeutic solutions forward with speed and precision.