Advanced Synthesis of Camptothecin-Steroid Conjugates for High-Potency Antitumor Drug Development
The pharmaceutical industry continuously seeks to optimize the therapeutic index of potent natural products, and Patent CN102746360B presents a groundbreaking approach through the synthesis of four novel camptothecin-steroid conjugates. Camptothecin (CPT), originally isolated from Camptotheca acuminata, is a renowned topoisomerase I inhibitor; however, its clinical application has been historically hindered by severe bladder toxicity, poor water solubility, and the instability of its E-ring lactone moiety in physiological conditions. This patent addresses these critical limitations by strategically esterifying the C20-hydroxyl group of camptothecin with various steroidal acids, creating hybrid molecules that leverage the lipophilic and membrane-permeable properties of steroids. The resulting conjugates, specifically designated as CPT-1 through CPT-4, demonstrate a remarkable enhancement in antitumor activity, with specific derivatives exhibiting potency significantly superior to the parent compound. For R&D directors and procurement specialists, this technology represents a viable pathway to developing next-generation oncology therapeutics with improved safety profiles and manufacturability.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional utilization of camptothecin in drug development has faced substantial biochemical and pharmacokinetic hurdles that limit its efficacy as a standalone therapeutic agent. The primary structural vulnerability lies in the E-ring α-hydroxy lactone, which is prone to hydrolytic ring-opening under physiological pH, converting the active lactone form into an inactive carboxylate form. Furthermore, the presence of an intramolecular hydrogen bond between the C20-hydroxyl and the adjacent ester carbonyl exacerbates this instability, leading to rapid degradation in vivo. Conventional attempts to modify the camptothecin scaffold often involve complex multi-step syntheses that require harsh reaction conditions, expensive protecting group strategies, or toxic transition metal catalysts, which complicate the purification process and introduce hazardous impurities. These factors collectively result in low overall yields, high production costs, and significant challenges in scaling up for commercial supply, making the reliable sourcing of high-purity camptothecin derivatives a persistent bottleneck for pharmaceutical manufacturers.
The Novel Approach
The innovative methodology disclosed in this patent circumvents these historical challenges by employing a direct esterification strategy at the C20 position using steroidal acids as the modifying agents. By attaching bulky, lipophilic steroid scaffolds—such as 3β-hydroxy-5-androstene or 4-aza-5α-androst-3-one derivatives—the synthesis effectively disrupts the detrimental intramolecular hydrogen bonding network. This structural modification not only stabilizes the critical lactone ring against hydrolysis but also dramatically enhances the molecule's lipophilicity, facilitating superior passive diffusion across cell membranes. 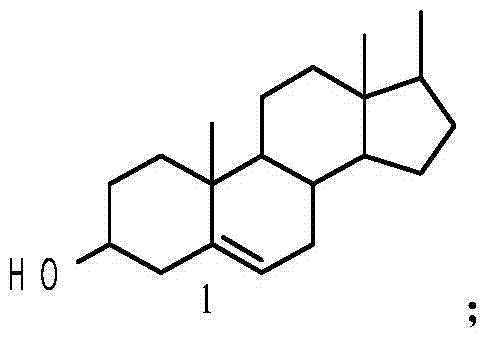 The use of diverse steroid backbones allows for fine-tuning of the biological activity, as evidenced by the varying potency of the four synthesized conjugates. This approach transforms a highly toxic, unstable natural product into a robust, high-performance pharmaceutical intermediate suitable for rigorous drug development pipelines, offering a distinct competitive advantage in the oncology sector.
The use of diverse steroid backbones allows for fine-tuning of the biological activity, as evidenced by the varying potency of the four synthesized conjugates. This approach transforms a highly toxic, unstable natural product into a robust, high-performance pharmaceutical intermediate suitable for rigorous drug development pipelines, offering a distinct competitive advantage in the oncology sector.
Mechanistic Insights into EDCI-DMAP Mediated Esterification
The core chemical transformation driving this synthesis is the carbodiimide-mediated coupling reaction, specifically utilizing 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl) in conjunction with 4-Dimethylaminopyridine (DMAP). In this mechanism, EDC acts as a dehydrating agent that activates the carboxylic acid group of the steroidal precursor, forming a highly reactive O-acylisourea intermediate. DMAP serves as a potent nucleophilic catalyst, attacking this intermediate to generate an even more reactive acylpyridinium species, which is subsequently attacked by the C20-hydroxyl group of camptothecin. This catalytic cycle proceeds efficiently at room temperature, avoiding the thermal stress that could otherwise degrade the sensitive camptothecin core. The reaction is conducted in anhydrous dichloromethane under an inert argon atmosphere, a critical parameter that prevents moisture-induced hydrolysis of the activated intermediates and ensures the integrity of the final ester linkage. This mechanistic pathway is highly atom-economical and generates urea byproducts that are easily removed during aqueous workup, ensuring a clean crude profile prior to purification.
Impurity control is paramount in the synthesis of complex conjugates intended for pharmaceutical use, and this protocol incorporates several built-in safeguards to minimize side reactions. The strict maintenance of anhydrous conditions prevents the hydrolysis of the EDC-activated acid back to the starting material, maximizing conversion efficiency. Furthermore, the use of a slight excess of steroidal acid and coupling reagents drives the equilibrium towards product formation without necessitating extreme temperatures that could promote racemization at the chiral C20 center. Post-reaction, the protocol specifies a rigorous purification sequence involving washing with dilute hydrochloric acid and saturated brine, followed by drying over anhydrous magnesium sulfate. The final purification via pressure column chromatography using a chloroform-methanol gradient ensures the removal of unreacted starting materials, urea byproducts, and any trace isomeric impurities, delivering a final product with the stringent purity specifications required for preclinical and clinical evaluation.
How to Synthesize Camptothecin-Steroid Conjugates Efficiently
The synthesis of these high-value conjugates follows a generalized yet precise protocol that can be adapted for various steroidal acids, providing a versatile platform for generating diverse libraries of anticancer agents. The process begins with the dissolution of the specific steroidal acid in anhydrous solvent, followed by the sequential addition of coupling agents and the camptothecin core under inert gas protection. The reaction is allowed to proceed at ambient temperature with continuous monitoring via Thin Layer Chromatography (TLC) to determine the optimal endpoint, preventing over-reaction or decomposition. Detailed standardized operating procedures for the synthesis of CPT-1, CPT-2, CPT-3, and CPT-4, including specific molar ratios and workup parameters, are outlined in the patent examples to ensure reproducibility and high yield.
- Dissolve the specific steroidal acid (e.g., 3β-hydroxy-5-androstene-17β-carboxylic acid) in anhydrous dichloromethane within a reaction vessel under inert atmosphere.
- Add coupling reagents EDC·HCl and catalytic DMAP to the solution, followed by the addition of Camptothecin (CPT) maintaining a precise molar ratio of 1: 3:3.6:6.
- Stir the reaction mixture at room temperature under argon protection, monitor progress via TLC, and purify the final conjugate using column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain and procurement perspective, the adoption of this EDCI-DMAP coupling technology offers substantial strategic benefits over traditional synthetic routes for camptothecin derivatives. The reliance on readily available steroidal acids as starting materials mitigates the risk of raw material shortages, as these compounds are widely produced commodities in the fine chemical industry. Furthermore, the reaction conditions are exceptionally mild, occurring at room temperature without the need for cryogenic cooling or high-pressure equipment, which translates to significantly reduced energy consumption and lower operational expenditures for manufacturing facilities. The elimination of toxic heavy metal catalysts, often required in cross-coupling reactions, removes the need for expensive and time-consuming metal scavenging steps, thereby streamlining the production timeline and reducing the environmental footprint of the manufacturing process.
- Cost Reduction in Manufacturing: The synthetic route described utilizes cost-effective reagents like EDC·HCl and DMAP, which are inexpensive compared to specialized organometallic catalysts. By avoiding complex protection-deprotection sequences and enabling a direct one-pot coupling strategy, the overall number of synthetic steps is minimized. This reduction in step count directly correlates to lower labor costs, reduced solvent usage, and decreased waste disposal fees, resulting in a more economically viable process for the large-scale production of high-purity pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: The robustness of the room-temperature reaction protocol ensures high batch-to-batch consistency, a critical factor for maintaining a stable supply of API intermediates. The use of common organic solvents like dichloromethane and standard purification techniques like column chromatography means that the process can be easily transferred between different manufacturing sites without requiring specialized infrastructure. This flexibility enhances supply chain resilience, allowing for rapid scale-up to meet fluctuating market demands for oncology drug candidates without compromising on quality or delivery timelines.
- Scalability and Environmental Compliance: The process generates minimal hazardous waste, primarily consisting of urea derivatives and aqueous salt solutions that are straightforward to treat. The absence of heavy metals simplifies regulatory compliance regarding residual impurities, accelerating the approval process for new drug applications. Additionally, the high selectivity of the EDCI-DMAP system reduces the formation of difficult-to-separate byproducts, improving the overall mass balance and yield, which is essential for sustainable commercial scale-up of complex pharmaceutical intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of these camptothecin-steroid conjugates, based on the detailed experimental data and background provided in the patent documentation. Understanding these aspects is crucial for stakeholders evaluating the feasibility of integrating these intermediates into their drug development pipelines.
Q: Why is esterification at the C20 position critical for Camptothecin stability?
A: Esterification at the C20-hydroxyl group eliminates the intramolecular hydrogen bond that typically destabilizes the E-ring lactone. This modification significantly enhances the stability of the active lactone form in vivo, thereby improving antitumor efficacy and reducing toxicity.
Q: What are the advantages of using EDCI-DMAP coupling for these conjugates?
A: The EDCI-DMAP system operates under mild room temperature conditions without requiring toxic heavy metal catalysts. This ensures a cleaner reaction profile, simplifies downstream purification, and aligns with green chemistry principles for scalable pharmaceutical manufacturing.
Q: How do steroid conjugates improve the pharmacological profile of Camptothecin?
A: Steroid moieties impart high lipophilicity and cell membrane affinity to the conjugate. This facilitates enhanced transmembrane penetration and potential receptor-mediated targeting, leading to significantly higher cytotoxicity against cancer cells like HepG2 compared to native Camptothecin.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Camptothecin-Steroid Conjugates Supplier
As a premier Contract Development and Manufacturing Organization (CDMO), NINGBO INNO PHARMCHEM possesses the technical expertise and infrastructure to bring these sophisticated camptothecin-steroid conjugates from laboratory scale to commercial reality. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the delicate balance of reaction parameters required for EDCI-DMAP coupling is maintained even at massive volumes. We operate state-of-the-art rigorous QC labs equipped to verify stringent purity specifications, guaranteeing that every batch of CPT-1, CPT-2, or other derivatives meets the exacting standards required for global pharmaceutical registration and clinical trials.
We invite pharmaceutical partners to collaborate with us to leverage this patented technology for your oncology portfolio. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our optimized manufacturing processes can accelerate your time-to-market while maximizing cost efficiency.