Advanced Alpha Nucleoside Synthesis: A Scalable Route for Antisense Drug Intermediates
Advanced Alpha Nucleoside Synthesis: A Scalable Route for Antisense Drug Intermediates
The rapid advancement of Antisense Technology has created an urgent demand for high-purity oligonucleotide monomers, specifically those with the alpha-configuration, which offer superior stability against enzymatic degradation compared to their natural beta-counterparts. Patent CN108424433B discloses a groundbreaking method for synthesizing these critical alpha nucleosides, addressing long-standing challenges in stereoselectivity and process universality. This innovative approach utilizes a streamlined three-step sequence involving the chlorination of protected ribofuranose, followed by coupling with various nucleobases and final deprotection. By shifting away from classical glycosylation methods that suffer from poor stereocontrol, this technology enables the efficient production of alpha-adenosine, alpha-guanosine, alpha-uridine, and alpha-cytidine. For pharmaceutical manufacturers, this represents a significant leap forward in securing reliable supply chains for next-generation antisense therapeutics, offering a robust pathway from laboratory scale to commercial manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of alpha nucleosides has been plagued by significant technical hurdles that impede commercial viability. One of the most common classical approaches involves the Silyl-Hilbert-Johnson reaction, where acyl-protected sugars are coupled with bases under Lewis acid catalysis. However, this method inherently favors the formation of the beta-isomer due to thermodynamic stability, resulting in a mixture where the desired alpha-epimer is the minor product. Separating these stereoisomers is notoriously difficult, often requiring extensive column chromatography or repeated crystallizations, which drastically reduces overall yield and increases production costs. Furthermore, alternative routes utilizing amino oxazole sugar compounds, while effective for uracil and cytosine derivatives, lack the necessary universality to synthesize purine-based alpha nucleosides like adenosine and guanosine efficiently.
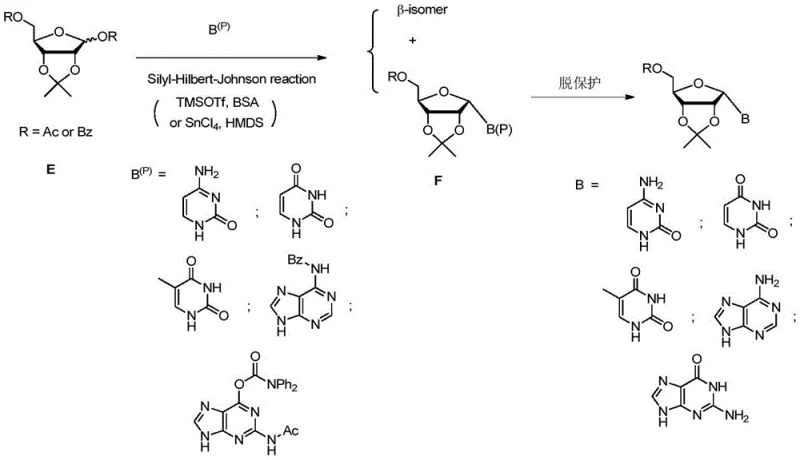
Another reported method relies on the cyclization of D-ribose with cyanamide to form key intermediates, but this pathway is similarly restricted in scope. The inability to apply a single, unified synthetic strategy across all four canonical nucleobases forces manufacturers to maintain multiple disparate production lines, complicating inventory management and quality control. The reliance on harsh conditions or specialized precursors in these legacy methods also introduces risks related to impurity profiles and batch-to-batch consistency. Consequently, the industry has faced a bottleneck in sourcing high-quality alpha nucleoside building blocks, limiting the speed at which new antisense drugs can progress from discovery to clinical trials and eventual market launch.
The Novel Approach
The methodology outlined in patent CN108424433B overcomes these historical barriers through a cleverly designed chlorination-coupling-deprotection sequence that prioritizes kinetic control and operational simplicity. By starting with 5-O-tert-butyldimethylsilane-2,3-O-isopropylidene-D-ribofuranose, the process leverages the steric and electronic properties of the protecting groups to direct the stereochemical outcome of the glycosidic bond formation. The key innovation lies in the initial chlorination step performed at cryogenic temperatures, which locks the sugar into the desired alpha-chloro configuration with exceptional selectivity. This intermediate is then directly coupled with the nucleobase in the presence of an inorganic base, avoiding the need for isolating sensitive halogenated sugars. The result is a versatile platform capable of producing a wide range of alpha nucleosides with high purity and minimal epimeric contamination.
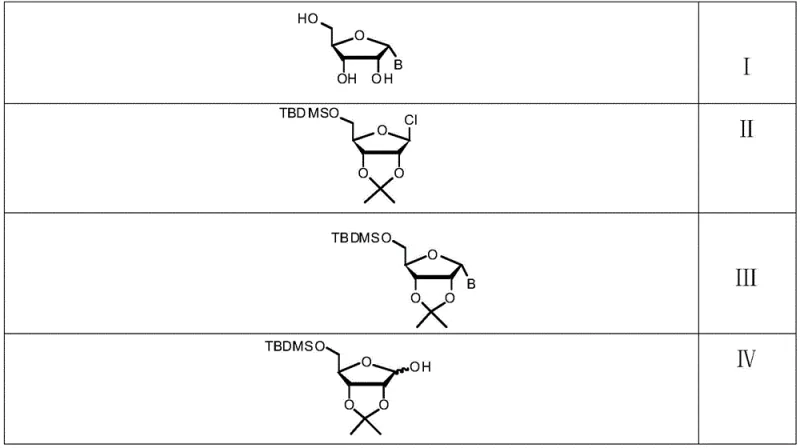
This novel approach not only solves the selectivity issue but also dramatically simplifies the workflow by allowing the use of crude reaction mixtures for subsequent steps. Unlike traditional routes that demand rigorous purification after every transformation, this method permits the telescoping of reactions, where the output of the chlorination step feeds directly into the coupling phase. This reduction in unit operations translates to lower solvent usage, reduced waste generation, and shorter cycle times. For supply chain managers, this means a more resilient production process that is less susceptible to delays caused by complex purification bottlenecks. The ability to synthesize both pyrimidine and purine alpha nucleosides using the same core protocol further enhances the economic attractiveness of this technology for large-scale API intermediate manufacturing.
Mechanistic Insights into Chlorination-Coupling Deprotection
The success of this synthesis hinges on the precise control of reaction conditions during the initial chlorination phase. The conversion of the hydroxyl group on the protected ribofuranose to a chloride is achieved using carbon tetrachloride and tris(dimethylamino)phosphine in tetrahydrofuran at temperatures ranging from -78 to -50°C. These cryogenic conditions are critical for suppressing anomerization and ensuring that the alpha-chloro sugar (Formula II) is formed with a selectivity ratio exceeding 20:1 over the beta-isomer. The use of tris(dimethylamino)phosphine acts as a promoter that facilitates the activation of the hydroxyl group without inducing the harsh acidic conditions that might lead to protecting group migration or sugar degradation. This mechanistic precision ensures that the stereochemical integrity established in the first step is preserved throughout the subsequent coupling reaction.
In the second stage, the crude alpha-chloro sugar reacts with the nucleobase in the presence of sodium hydride in acetonitrile. Sodium hydride serves as a strong, non-nucleophilic base that deprotonates the nitrogen atom of the heterocyclic base, generating a nucleophile capable of displacing the chloride leaving group. The reaction proceeds via an SN2-like mechanism where the configuration at the anomeric center is inverted or retained depending on the specific neighboring group participation, but in this optimized system, the conditions favor the formation of the alpha-N-glycosidic bond. Finally, the deprotection step utilizes a strongly acidic ion exchange resin in a mixture of acetonitrile and water. This heterogeneous catalysis offers a distinct advantage over homogeneous acids, as the resin can be simply filtered off, eliminating the need for neutralization and extraction steps that often lead to product loss. This mild yet effective deprotection strategy ensures the final alpha nucleoside is obtained with high chemical purity and minimal structural damage.
How to Synthesize Alpha Nucleosides Efficiently
The synthesis of alpha nucleosides via this patented route is designed for operational efficiency, minimizing the need for intermediate isolation and complex workups. The process begins with the preparation of the alpha-chloro sugar intermediate, which is generated in situ and used directly in the coupling reaction without purification. This telescoping strategy significantly reduces processing time and solvent consumption. Following the coupling with the chosen nucleobase, the protected nucleoside is subjected to acid-catalyzed deprotection using a solid resin catalyst, which simplifies the removal of acidic species. Detailed standardized synthesis steps, including specific molar ratios, temperature controls, and quenching procedures for alpha-adenosine and alpha-guanosine, are provided in the technical guide below to ensure reproducible results in your facility.
- Chlorinate 5-O-tert-butyldimethylsilane-2,3-O-isopropylidene-D-ribofuranose using carbon tetrachloride and tris(dimethylamino)phosphine at -78 to -50°C to form the alpha-chloro sugar intermediate.
- Couple the crude chloro sugar intermediate with a nucleobase (Adenine, Guanine, Cytosine, Uracil, or Thymine derivatives) using sodium hydride in acetonitrile.
- Deprotect the coupled intermediate using a strong acidic ion exchange resin catalyst in acetonitrile and water to yield the final alpha nucleoside.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement specialists and supply chain directors, the adoption of this synthesis method offers tangible benefits that extend beyond mere technical feasibility. The primary advantage lies in the drastic simplification of the manufacturing workflow, which directly correlates to reduced operational expenditures. By eliminating the need to isolate and purify the chlorinated sugar intermediate and the protected nucleoside precursor, the process removes two major cost centers associated with chromatography and crystallization. This reduction in unit operations not only lowers the cost of goods sold but also decreases the physical footprint required for production, allowing for higher throughput within existing infrastructure. Furthermore, the use of commercially available starting materials and common reagents like sodium hydride and carbon tetrachloride ensures that raw material sourcing remains stable and cost-effective, mitigating the risk of supply disruptions.
- Cost Reduction in Manufacturing: The elimination of intermediate purification steps is a game-changer for cost optimization. Traditional methods often require multiple column chromatography separations to remove beta-isomers and side products, which consumes vast amounts of silica gel and solvents. In contrast, this novel route allows for the direct use of crude reaction liquids, meaning that expensive purification resources are reserved solely for the final product. Additionally, the replacement of homogeneous acid catalysts with solid acidic ion exchange resins removes the need for complex neutralization and washing procedures, further reducing labor and utility costs. The overall yield improvement resulting from higher stereoselectivity also means less starting material is wasted, contributing to a leaner and more economical production model.
- Enhanced Supply Chain Reliability: The universality of this synthetic platform ensures that a single production line can be adapted to manufacture various alpha nucleosides, from adenosine to uridine, with minimal retooling. This flexibility is crucial for maintaining supply continuity in a market where demand for specific antisense therapeutics can fluctuate rapidly. Since the process relies on robust, scalable chemistry rather than finicky enzymatic or highly specialized organometallic reactions, the risk of batch failure is significantly minimized. The use of standard industrial solvents like acetonitrile and tetrahydrofuran, along with readily available inorganic bases, ensures that the supply chain is not dependent on exotic or single-source reagents, thereby enhancing the resilience of the procurement strategy against global market volatility.
- Scalability and Environmental Compliance: Scaling this process from laboratory to commercial tonnage is straightforward due to the absence of sensitive transition metal catalysts that often require stringent removal protocols to meet regulatory limits. The heterogeneous nature of the deprotection step facilitates easy catalyst recovery and reuse, aligning with green chemistry principles and reducing hazardous waste generation. The simplified workup procedures, which primarily involve extraction and filtration rather than distillation or complex chromatography, lower the energy intensity of the manufacturing process. This environmental efficiency not only reduces disposal costs but also positions the manufacturer favorably regarding increasingly strict environmental regulations, ensuring long-term operational sustainability and compliance with international pharmaceutical standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this alpha nucleoside synthesis technology. These answers are derived directly from the experimental data and process descriptions found in the patent literature, providing a clear understanding of the method's capabilities and limitations. Understanding these details is essential for R&D teams evaluating the feasibility of integrating this route into their existing pipelines and for procurement officers assessing the quality and consistency of the resulting intermediates.
Q: How does this method improve alpha-selectivity compared to traditional Silyl-Hilbert-Johnson reactions?
A: Traditional Silyl-Hilbert-Johnson reactions often favor the beta-configuration, requiring difficult separation of epimers. This novel method utilizes a specific low-temperature chlorination step (-78 to -50°C) that achieves an alpha-to-beta selectivity ratio greater than 20:1, significantly simplifying downstream purification.
Q: Can this synthesis route be applied to all major nucleobases?
A: Yes, unlike previous methods limited to uracil and cytosine derivatives, this process demonstrates strong universality. It successfully synthesizes alpha-adenosine, alpha-guanosine, alpha-uridine, and alpha-cytidine using the same core chlorination and coupling strategy.
Q: What are the purification advantages of using crude intermediates?
A: The process allows the direct use of reaction liquids containing intermediates (Formula II and III) without isolation. This eliminates multiple column chromatography or crystallization steps for intermediates, reducing solvent consumption, processing time, and overall material loss before the final purification.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Alpha Nucleoside Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-purity alpha nucleosides play in the development of cutting-edge antisense therapies. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the rigorous demands of global pharmaceutical clients. We are committed to delivering intermediates with stringent purity specifications, utilizing our rigorous QC labs to verify stereochemical integrity and impurity profiles for every batch. Our capability to implement complex synthetic routes like the chlorination-coupling-deprotection method allows us to offer a stable and high-quality supply of alpha-adenosine, alpha-guanosine, and other critical nucleoside analogs.
We invite you to collaborate with us to optimize your supply chain for antisense drug manufacturing. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our advanced synthesis capabilities can accelerate your project timelines and reduce your overall manufacturing costs. Let us be your partner in bringing the next generation of genetic medicines to market.