Optimizing ADC Payload Production: A Technical Analysis of Patent CN109963835B
Introduction to Advanced ADC Payload Synthesis
The landscape of Antibody-Drug Conjugate (ADC) manufacturing is constantly evolving, driven by the need for more efficient and scalable synthesis of cytotoxic payloads. Patent CN109963835B, published in late 2022, introduces a groundbreaking preparation method for a novel toxin and its key intermediates, specifically focusing on compounds of general Formula (I). These structures are critical components in the construction of ligand-cytotoxic drug conjugates, which have shown immense promise in oncology treatments by regulating receptors to combat cancer. The patent details a sophisticated chemical architecture that integrates a maleimide linker for antibody conjugation with a complex peptide-based cytotoxic warhead. By addressing the limitations of previous synthetic routes, this technology offers a robust pathway for producing high-purity intermediates essential for next-generation therapeutics. The structural complexity of Formula (I), featuring multiple chiral centers and sensitive functional groups, demands a synthesis strategy that balances reactivity with stereochemical integrity.
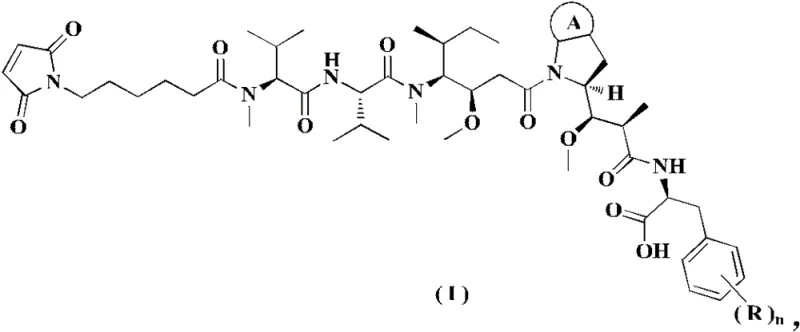
For procurement and supply chain leaders, understanding the nuances of this patent is vital. It represents not just a chemical improvement, but a strategic advantage in securing reliable sources for high-value pharmaceutical intermediates. The ability to synthesize these complex molecules with fewer steps and higher yields directly translates to reduced manufacturing costs and improved supply continuity. As the demand for ADCs grows globally, the methodologies outlined in CN109963835B provide a blueprint for industrial-scale production that mitigates the risks associated with long, low-yielding synthetic sequences. This report delves deep into the technical merits of this invention, analyzing how it reshapes the economics of ADC payload manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this innovation, the synthesis of similar MMAF analogs relied on linear or less efficient convergent strategies, such as those disclosed in patent application WO2016127790A1. In these conventional routes, the assembly of the final molecule often required a direct condensation of large fragments followed by multiple subsequent modifications. Specifically, the prior art typically involved a four-step sequence to achieve the final coupling and deprotection necessary to reveal the active pharmacophore. This elongated process inherently suffers from cumulative yield losses, where each additional purification and isolation step erodes the overall material throughput. Furthermore, handling sensitive intermediates over multiple steps increases the risk of epimerization at chiral centers and degradation of the maleimide moiety, which is susceptible to hydrolysis or ring-opening under prolonged reaction conditions. The reliance on such multi-step protocols creates bottlenecks in production, leading to higher consumption of solvents, reagents, and labor hours, ultimately inflating the cost of goods sold (COGS) for the final API.
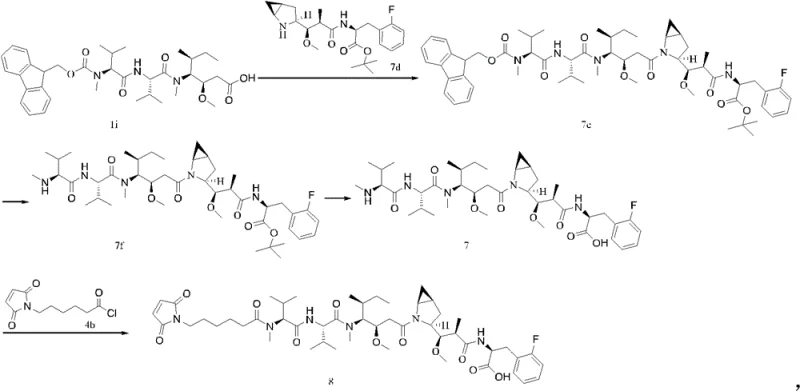
The Novel Approach
In stark contrast, the methodology presented in CN109963835B employs a highly optimized convergent strategy that drastically streamlines the synthesis. The core innovation lies in the preparation of a specific intermediate of Formula (III), which is then coupled with a complementary fragment of Formula (IV) to directly access the protected precursor of the target molecule. This approach effectively collapses the critical final assembly into just two major steps: a condensation reaction to form the amide bond, followed by a single deprotection event to unveil the free carboxylic acid. By pre-functionalizing the fragments with orthogonal protecting groups, such as the 2,4-dimethoxybenzyl (DMB) group, the chemists can control the reactivity with precision. This reduction from four steps to two in the key coupling phase is not merely a numerical improvement; it represents a fundamental shift in process efficiency. The novel route minimizes the exposure of sensitive intermediates to harsh conditions, thereby preserving optical purity and chemical integrity. For a reliable pharmaceutical intermediate supplier, adopting this shorter route means faster turnaround times and a more robust process capable of withstanding the rigors of commercial scale-up.
Mechanistic Insights into Convergent Amide Coupling
The success of this synthetic strategy hinges on the precise execution of amide bond formation between the carboxyl-terminated Fragment III and the amine-terminated Fragment IV. The patent specifies the use of advanced coupling reagents, with a strong preference for 2-(7-azobenzotriazole)-N,N,N',N'-tetramethylurea hexafluorophosphate (HATU). HATU is renowned for its ability to activate carboxylic acids rapidly while minimizing racemization, a critical factor when dealing with the multiple chiral amino acid residues present in the peptide backbone of Formula (III). The reaction is typically conducted in a mixed solvent system of dichloromethane and N,N-dimethylformamide (DMF), which ensures solubility of the bulky peptide fragments. The presence of a non-nucleophilic base, such as N,N-diisopropylethylamine (DIPEA), is essential to scavenge the acid generated during activation and to maintain the amine nucleophile in its reactive free-base form. This carefully balanced environment facilitates the formation of the activated ester intermediate, which is then attacked by the amine of Fragment IV to forge the peptide bond with high fidelity.
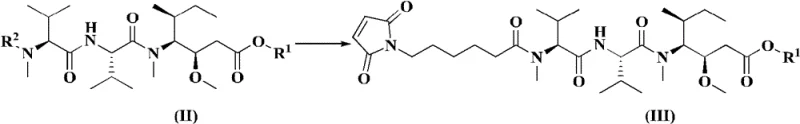
Beyond the coupling itself, the management of protecting groups is a masterclass in orthogonal chemistry. The synthesis utilizes the Fmoc (9-fluorenylmethoxycarbonyl) group for amine protection, which can be cleanly removed under basic conditions using secondary amines like diethylamine without affecting other sensitive functionalities. Conversely, the carboxyl groups are masked with DMB or similar benzyl-type protectors that are stable to base but cleavable under acidic conditions, such as trifluoroacetic acid (TFA). This orthogonality allows for the sequential assembly of the molecule without cross-reactivity. For instance, the intermediate Formula (IIIa) can be deprotected to Formula (IIIb) selectively, enabling it to participate in the final coupling. The final deprotection step uses TFA to remove the DMB group, revealing the free carboxylic acid required for subsequent conjugation to the antibody linker. This level of control over impurity profiles is paramount for R&D directors, as it ensures that the final product meets the stringent purity specifications required for clinical applications, reducing the burden on downstream purification processes.
How to Synthesize MMAF Analog Intermediate Efficiently
Implementing this synthesis requires a disciplined approach to reaction monitoring and purification. The process begins with the preparation of the maleimide-containing fragment, where 6-maleimidohexanoic acid is coupled to the peptide chain under mild conditions to prevent maleimide ring opening. Following this, the fragments are purified via silica gel column chromatography using gradients of dichloromethane and methanol or ethyl acetate to remove urea byproducts from the coupling reagents. The final coupling step is exothermic and requires temperature control, typically maintained between 20°C and 30°C, to ensure consistent reaction kinetics. Detailed standardized synthetic steps see the guide below.
- Prepare the carboxyl-protected fragment (Formula III) via amidation of Formula II with 6-maleimidohexanoic acid.
- Couple Formula III with the amine fragment (Formula IV) using HATU and DIPEA to form the protected intermediate (Formula V).
- Perform final deprotection using trifluoroacetic acid to yield the target Formula I compound.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the technical improvements outlined in CN109963835B translate directly into tangible supply chain benefits. The reduction in synthetic steps eliminates the need for multiple isolation and purification stages, which are often the most resource-intensive parts of chemical manufacturing. This streamlining leads to substantial cost savings in terms of solvent usage, waste disposal, and labor. Furthermore, the improved yield means that less starting material is required to produce the same amount of final product, optimizing the utilization of expensive chiral building blocks. For procurement managers, this efficiency offers a buffer against raw material price volatility and ensures a more stable cost structure for long-term contracts.
- Cost Reduction in Manufacturing: The elimination of two full synthetic steps in the critical coupling phase drastically reduces the operational expenditure associated with the production of this intermediate. By avoiding the isolation of unstable intermediates that were necessary in the prior art, the process minimizes material loss and reduces the consumption of high-purity solvents and chromatography media. This leaner manufacturing footprint allows for a significant reduction in the overall cost of goods, making the final ADC therapy more economically viable without compromising on quality standards.
- Enhanced Supply Chain Reliability: Shorter synthetic routes are inherently more robust and less prone to failure. Each chemical step introduces a potential point of delay or deviation; by cutting the step count, the probability of batch failures is significantly lowered. This reliability is crucial for maintaining continuous supply to downstream conjugation facilities. Additionally, the use of commercially available and stable protecting group reagents ensures that the supply chain is not dependent on exotic or hard-to-source specialty chemicals, further securing the production timeline against external disruptions.
- Scalability and Environmental Compliance: The simplified process is highly amenable to scale-up from kilogram to tonne levels. Fewer unit operations mean smaller equipment footprints and reduced energy consumption for heating, cooling, and drying. Moreover, the reduction in solvent volume and waste generation aligns with modern green chemistry principles and environmental regulations. This makes the technology not only economically attractive but also environmentally sustainable, facilitating easier regulatory approval and community acceptance for manufacturing sites.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis method. These insights are derived directly from the experimental data and claims within the patent documentation, providing clarity on the practical aspects of adopting this technology for industrial production.
Q: How does the new synthesis method improve upon prior art like WO2016127790A1?
A: The new method reduces the key coupling sequence from four steps to just two steps, significantly minimizing purification losses and operational time while improving overall yield.
Q: What are the critical protecting groups used in this synthetic route?
A: The process utilizes orthogonal protecting groups such as Fmoc (9-fluorenylmethoxycarbonyl) for amines and DMB (2,4-dimethoxybenzyl) for carboxyls, allowing for selective deprotection under mild conditions.
Q: Why is this intermediate important for Antibody-Drug Conjugates (ADCs)?
A: This compound serves as a potent cytotoxic payload (MMAF analog) that can be conjugated to antibodies via the maleimide linker, targeting specific cancer cells with high precision.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable MMAF Analog Supplier
The technical potential of Patent CN109963835B is immense, offering a pathway to high-quality ADC payloads that meet the rigorous demands of the biopharmaceutical industry. At NINGBO INNO PHARMCHEM, we possess the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring such complex molecules to market. Our state-of-the-art facilities are equipped with stringent purity specifications and rigorous QC labs to ensure that every batch of intermediate produced adheres to the highest global standards. We understand the critical nature of these materials in the drug development timeline and are committed to delivering consistency and excellence.
We invite you to collaborate with us to leverage this advanced synthetic technology for your ADC programs. Our team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this optimized route can enhance your project's economics. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us partner with you to accelerate your therapeutic solutions.