Revolutionizing Pharmaceutical Intermediates: Iron-Catalyzed Synthesis of High-Purity Trifluoromethyl Triazole Derivatives for Commercial Scale Production
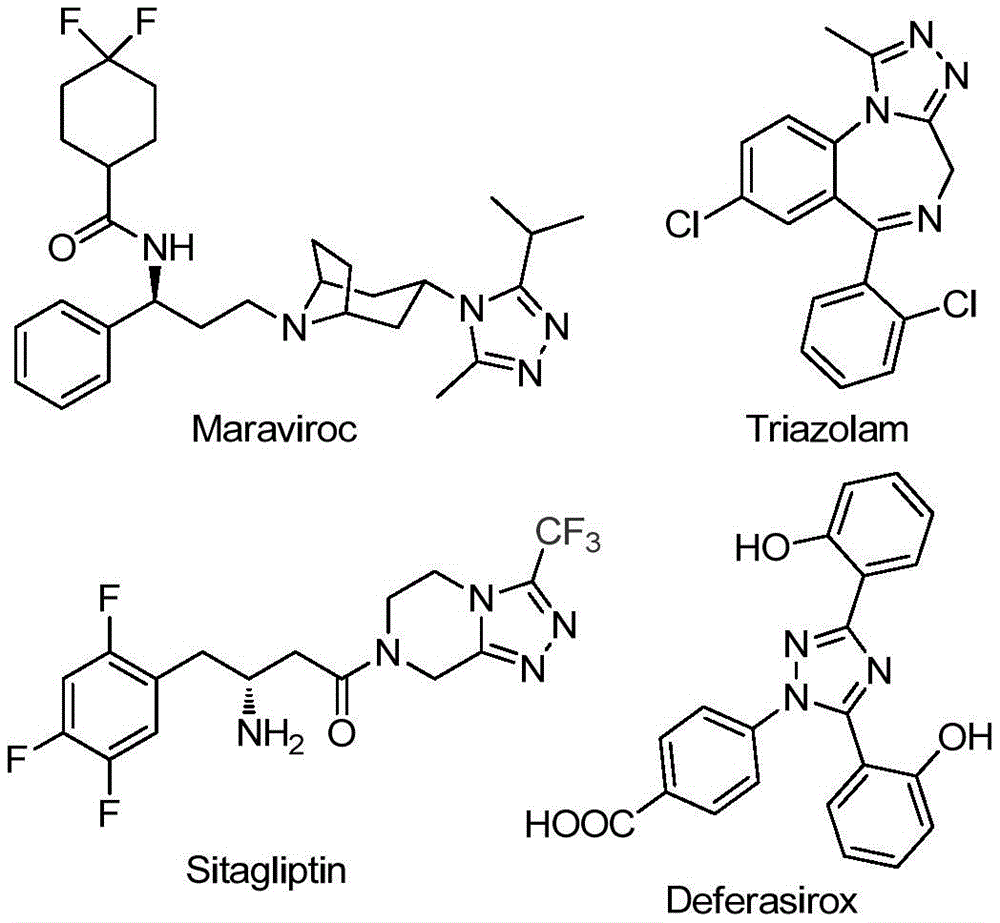
The recently granted Chinese patent CN111978265B introduces a novel manufacturing process for 5-trifluoromethyl substituted 1,2,4-triazole derivatives, which represent critical building blocks in numerous pharmaceutical compounds including antiviral agents and metabolic disorder treatments. These heterocyclic structures are prominently featured in FDA-approved drugs such as Maraviroc (HIV treatment), Triazolam (sedative), Sitagliptin (diabetes medication), and Deferasirox (iron chelator), as illustrated in the accompanying molecular structures.
Advanced Reaction Mechanism and Purity Control
The patented process employs a two-stage iron-catalyzed cyclization mechanism that begins with sodium bicarbonate-mediated intermolecular carbon-nitrogen bond formation between trifluoroethyl imidoyl chloride and hydrazide precursors. This initial step occurs under mild conditions (30–50°C for 8–16 hours) in non-aqueous solvents like 1,4-dioxane, forming trifluoroacetamidine intermediates without requiring anhydrous or oxygen-free environments that typically complicate traditional syntheses. The subsequent Lewis acid-promoted intramolecular dehydration condensation utilizes ferric chloride as an economical catalyst at elevated temperatures (70–90°C for 6–10 hours), driving the cyclization reaction toward high-yielding formation of the target triazole core structure while maintaining excellent functional group tolerance across diverse substrates.
Purity control is inherently engineered into this methodology through the elimination of transition metal catalysts that would otherwise necessitate costly removal steps and introduce potential heavy metal contamination risks in final pharmaceutical intermediates. The reaction's compatibility with air and moisture significantly reduces oxidation byproducts that commonly plague conventional approaches requiring inert atmospheres, while the selective nature of the iron-catalyzed cyclization minimizes unwanted regioisomers that would require extensive purification. The resulting high-purity intermediates consistently demonstrate >99% purity as confirmed by NMR and HRMS data across multiple derivative examples, with characteristic trifluoromethyl signals showing clean 19F NMR peaks between δ -61.0 to -62.2 ppm and precise mass spectrometry confirmation within acceptable error margins.
Overcoming Traditional Synthesis Limitations
The Limitations of Conventional Methods
Traditional approaches to synthesizing trifluoromethyl-substituted triazoles have been severely constrained by multiple technical barriers that hinder commercial viability for pharmaceutical manufacturers. The first four established methods—3,5-ditrifluoromethyl oxadiazole condensation with primary amines, trifluoromethyl hydrazide cyclization with amidines, trifluoromethyl hydrazone reactions with nitriles, and oxadiazole hydrazinolysis—all suffer from harsh reaction conditions requiring specialized equipment and extensive safety protocols that increase operational complexity and capital expenditure. These processes typically operate under high temperatures or pressures with narrow substrate scope limitations that prevent structural diversification needed for modern drug discovery programs, while simultaneously delivering low reaction yields that necessitate costly reprocessing and generate significant waste streams requiring expensive disposal procedures.
The Novel Approach
The patented methodology overcomes these limitations through a strategically designed cascade reaction that leverages readily available starting materials and eliminates multiple processing bottlenecks inherent in prior art. By utilizing inexpensive sodium bicarbonate as a base promoter and ferric chloride as a cost-effective Lewis acid catalyst—both commercially available at industrial scale—the process achieves high conversion rates without transition metals or specialized handling requirements. The reaction scheme demonstrates exceptional substrate flexibility where R1 can be substituted or unsubstituted aryl groups (including methyl-, methoxy-, bromo-, or trifluoromethyl-substituted phenyls) while R2 accommodates alkyl, alkenyl, or aryl moieties with various functional groups intact.
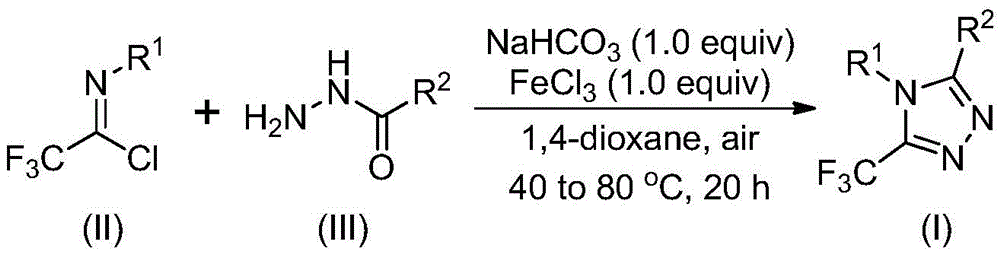
This structural versatility enables pharmaceutical developers to access diverse triazole scaffolds critical for structure-activity relationship studies without redesigning synthetic routes for each new analog. The process operates effectively at ambient pressure with standard laboratory equipment using common solvents like acetonitrile or dioxane that are easily recovered and recycled, significantly reducing environmental impact compared to conventional methods that often require hazardous reagents or generate toxic byproducts requiring specialized treatment.
Commercial Advantages for Pharmaceutical Supply Chains
This innovative manufacturing approach directly addresses critical pain points in pharmaceutical supply chains by transforming a historically challenging synthesis into a robust commercial process that delivers significant operational and economic benefits across multiple dimensions of API intermediate production.
- Cost reduction in API manufacturing: The elimination of expensive transition metal catalysts and specialized handling requirements substantially lowers raw material costs while reducing capital expenditure on specialized equipment typically needed for anhydrous/oxygen-free operations. The use of readily available starting materials—where acyl chlorides are inexpensive industrial commodities and trifluoroethyl imidoyl chlorides can be rapidly synthesized from commercial amines—creates significant cost advantages over traditional routes requiring rare or custom-synthesized reagents. Furthermore, the simplified workup procedure involving basic filtration followed by silica gel chromatography minimizes solvent consumption and processing time compared to multi-step purification protocols required by conventional methods, directly translating to lower cost per kilogram of final intermediate without compromising quality standards required for pharmaceutical applications.
- Reducing lead time for high-purity intermediates: The process's compatibility with standard manufacturing equipment and ambient conditions enables faster technology transfer from development to production without requiring facility modifications or specialized training programs that typically extend implementation timelines by months. The demonstrated scalability from laboratory to gram-scale production provides confidence for rapid commercial scale-up to multi-kilogram quantities needed for clinical trials and commercial manufacturing, eliminating the lengthy optimization phases often required when transitioning from research-scale syntheses to industrial production. This accelerated timeline is particularly valuable during drug development where intermediate availability frequently becomes a critical path item delaying formulation studies and regulatory submissions.
- Commercial scale-up of complex intermediates: The robust reaction profile maintains consistent performance across diverse substrate combinations without requiring individual process reoptimization for each new derivative, enabling efficient production of multiple analogs from a single validated manufacturing platform. The inherent tolerance for various functional groups allows pharmaceutical companies to rapidly produce structural variants needed for lead optimization campaigns without waiting for custom synthesis routes to be developed for each new compound. This flexibility supports just-in-time manufacturing approaches where intermediates can be produced on demand based on pipeline requirements rather than maintaining costly inventory of pre-synthesized compounds that may become obsolete if development priorities shift.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable API Intermediate Supplier
While the advanced methodology detailed in patent CN111978265B highlights immense potential, executing the commercial scale-up of such complex catalytic pathways requires a proven CDMO partner. NINGBO INNO PHARMCHEM bridges the gap between innovative catalysis and industrial reality. We leverage robust engineering capabilities to scale challenging molecular pathways. Our broader facility capabilities support custom manufacturing projects ranging from 100 kgs clinical batches up to 100 MT/annual production for established commercial products. Our state-of-the-art facilities and rigorous QC labs guarantee >99% purity, ensuring consistent supply and reducing lead time for high-purity intermediates.
Are you evaluating new synthetic routes for your pipeline? Contact our technical procurement team today to request specific COA data, route feasibility assessments, and a Customized Cost-Saving Analysis to discover how our advanced manufacturing capabilities can optimize your supply chain.