Scalable Synthesis of 1-(Chloromethyl)bicyclo[1.1.1]pentane for Advanced Pharmaceutical Intermediates
Introduction to Patent CN114591137B
The pharmaceutical industry's relentless pursuit of novel bioisosteres has placed bicyclo[1.1.1]pentane (BCP) derivatives at the forefront of medicinal chemistry innovation. These unique cage-like structures serve as exceptional substitutes for aromatic rings and tert-butyl groups, offering improved pharmacokinetic profiles while maintaining biological activity. However, the widespread adoption of these valuable scaffolds has historically been hindered by complex synthesis routes and harsh reaction conditions. Patent CN114591137B introduces a groundbreaking synthetic methodology that addresses these critical bottlenecks, providing a robust pathway for the preparation of halogenated methyl propellane derivatives. This technology represents a significant leap forward for any organization seeking a reliable pharmaceutical intermediate supplier capable of delivering high-quality BCP building blocks.
The core innovation lies in a streamlined three-step sequence that bypasses the need for expensive transition metal catalysts and specialized photochemical equipment. By leveraging a radical-based approach starting from accessible bromide precursors, the method achieves high purity and substantial yields under relatively mild thermal conditions. For R&D directors and process chemists, this translates to a more predictable and controllable synthesis platform. The ability to produce 1-(chloromethyl)bicyclo[1.1.1]pentane and its derivatives efficiently opens new avenues for drug discovery programs that rely on these rigid, three-dimensional motifs to enhance solubility and metabolic stability.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of substituted bicyclo[1.1.1]pentane derivatives has been fraught with challenges that impede commercial viability. Traditional routes often rely on multi-step sequences involving sensitive organometallic reagents or harsh reduction conditions. For instance, earlier literature describes pathways starting from bicyclo[1.1.1]pentane carboxylic acid, which requires reduction with lithium aluminum hydride followed by sulfonation and subsequent substitution. These methods are not only lengthy but also involve hazardous reagents and difficult purification steps that lower overall throughput.
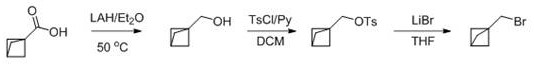
Furthermore, alternative approaches utilizing photocatalysis have emerged, employing fac-Ir(ppy)3 and blue light irradiation to achieve bromoalkylation. While chemically elegant, these photochemical methods introduce significant engineering hurdles for large-scale manufacturing. The requirement for specialized light sources and the use of costly iridium catalysts create economic barriers that make cost reduction in API manufacturing difficult to achieve. Additionally, the separation of trace metal impurities from the final product adds another layer of complexity to the downstream processing, often necessitating extensive chromatography or scavenging protocols.
The Novel Approach
In stark contrast, the methodology disclosed in CN114591137B offers a direct and efficient alternative that eliminates the need for light irradiation and precious metal catalysts. The process begins with the formation of tricyclo[1.1.1.0]pentane (TCP) from a dibromodichloro precursor using methyllithium, followed by a radical addition of iodomethyl chloride. This sequence effectively constructs the BCP core and installs the functional handle in a controlled manner. The final step involves a radical reduction to remove the iodine atom, yielding the target chloromethyl derivative with high fidelity.
![Novel synthetic route for 1-(chloromethyl)bicyclo[1.1.1]pentane showing high efficiency](/insights/img/chloromethyl-bcp-synthesis-pharma-supplier-20260308095748-013.png)
This novel approach drastically simplifies the operational landscape for chemical production. By avoiding photochemical reactors, manufacturers can utilize standard stainless steel vessels equipped with conventional heating and cooling systems. The reaction conditions are mild, typically proceeding at temperatures ranging from -45°C to 110°C depending on the specific step, which is well within the capabilities of standard pilot and production plants. This shift from specialized photochemistry to thermal radical chemistry significantly lowers the barrier to entry for commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Radical-Mediated BCP Functionalization
The success of this synthetic strategy hinges on the precise control of radical intermediates during the functionalization of the strained propellane system. In the second step of the process, the highly strained central bond of the tricyclo[1.1.1.0]pentane undergoes homolytic cleavage upon interaction with the iodomethyl radical species. This addition relieves the ring strain and generates a stabilized bridgehead radical, which subsequently captures an iodine atom to form the 1-(chloromethyl)-3-iodobicyclo[1.1.1]pentane intermediate. Understanding this mechanism is crucial for optimizing reaction times and minimizing side products such as oligomers or over-halogenated species.
Impurity control is further enhanced in the final reduction step, where initiators like AIBN or BPO generate radicals to abstract the iodine atom from the intermediate. The use of tin or silicon-based hydrides ensures a clean hydrogen atom transfer, regenerating the bridgehead radical which then abstracts a hydrogen to complete the cycle. This mechanism allows for the production of high-purity bicyclo[1.1.1]pentane derivatives, with the patent reporting purities exceeding 99%. The ability to crystallize intermediates or distill final products directly from the reaction mixture underscores the robustness of this chemical design against impurity formation.
How to Synthesize 1-(Chloromethyl)bicyclo[1.1.1]pentane Efficiently
Implementing this synthesis requires careful attention to temperature control and reagent stoichiometry, particularly during the initial organolithium step. The process is designed to be flexible, allowing for both stepwise and one-pot procedures depending on the specific infrastructure available. Detailed standardized operating procedures regarding solvent selection, such as the use of methyl tert-butyl ether or n-pentane, are critical for maximizing yield and safety. For a comprehensive guide on executing this transformation, please refer to the standardized protocol below.
- React the dibromodichloro starting material with methyllithium at low temperatures (-45°C to -15°C) to generate tricyclo[1.1.1.0]pentane (TCP).
- Subject the generated TCP to a radical reaction with iodomethyl chloride (CH2ClI) to form the 1-(chloromethyl)-3-iodobicyclo[1.1.1]pentane intermediate.
- Reduce the iodo-intermediate using a radical reducing agent (such as tributyltin hydride or triethylsilane) and an initiator (AIBN or BPO) to yield the final chloromethyl derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the transition to this patented synthesis method offers tangible benefits that extend beyond mere chemical elegance. The elimination of expensive iridium photocatalysts and the avoidance of specialized lighting equipment directly translate to reduced capital expenditure and lower raw material costs. Furthermore, the use of commodity chemicals like methyllithium, iodomethyl chloride, and common radical initiators ensures a stable and resilient supply chain, mitigating the risks associated with sourcing exotic reagents.
- Cost Reduction in Manufacturing: The removal of precious metal catalysts eliminates the need for costly metal scavenging steps and reduces the overall bill of materials. By utilizing thermal initiation instead of photochemical activation, energy consumption profiles become more predictable and manageable, leading to substantial cost savings in utility usage. The high yields reported, often exceeding 80%, mean less raw material waste and higher throughput per batch, optimizing the cost per kilogram of the final active pharmaceutical ingredient intermediate.
- Enhanced Supply Chain Reliability: The reliance on widely available industrial solvents and reagents ensures that production schedules are not disrupted by the scarcity of niche chemicals. The robustness of the reaction conditions allows for flexibility in sourcing, as the process is not sensitive to minor variations in reagent grade that might plague more sensitive catalytic systems. This reliability is essential for maintaining continuous supply lines to downstream drug substance manufacturers who demand consistent quality and availability.
- Scalability and Environmental Compliance: The process is inherently scalable, moving seamlessly from gram-scale laboratory optimization to multi-ton commercial production without the need for reactor redesign. The absence of heavy metal photocatalysts simplifies waste stream management and environmental compliance, reducing the burden on wastewater treatment facilities. Additionally, the ability to perform the reaction in standard vessels facilitates rapid technology transfer between sites, ensuring global supply continuity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on performance metrics and operational requirements. Understanding these details is vital for making informed decisions about adopting this route for your specific project needs.
Q: How does this synthesis method improve upon traditional photocatalytic routes?
A: Unlike traditional methods that require expensive iridium photocatalysts and specific blue light irradiation, this patented process operates under thermal conditions without light, significantly simplifying reactor requirements and reducing catalyst costs for large-scale production.
Q: What yields can be expected from this synthetic route?
A: The patent data indicates that the synthesis of chloromethyl BCP derivatives can achieve yields exceeding 80%, with specific solution-phase preparations reaching nearly 95% yield as measured by internal standard methods.
Q: Is this process suitable for industrial scale-up?
A: Yes, the process utilizes mild reaction conditions, avoids high-pressure mercury lamps, and employs readily available reagents like methyllithium and AIBN, making it highly amenable to commercial scale-up from kilogram to tonnage levels.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 1-(Chloromethyl)bicyclo[1.1.1]pentane Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of bicyclo[1.1.1]pentane derivatives in modern drug design. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from benchtop discovery to market-ready supply. Our state-of-the-art facilities are equipped to handle the specific thermal and safety requirements of radical chemistry, guaranteeing stringent purity specifications and rigorous QC labs testing for every batch we produce.
We invite you to collaborate with us to leverage this advanced synthetic technology for your next generation of therapeutics. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our expertise can accelerate your development timeline and optimize your manufacturing economics.