Advanced Metal-Free Synthesis of Alpha-Trifluoromethyl-Gamma-Carbonyl Esters for Commercial Scale-Up
Advanced Metal-Free Synthesis of Alpha-Trifluoromethyl-Gamma-Carbonyl Esters for Commercial Scale-Up
The introduction of fluorine-containing groups, particularly the trifluoromethyl moiety, into organic molecules has become a cornerstone strategy in modern medicinal chemistry and agrochemical development. As illustrated by blockbuster pharmaceuticals such as Prozac® and Efavirenz, the presence of a trifluoromethyl group can drastically alter the metabolic stability, lipophilicity, and binding affinity of a drug candidate. 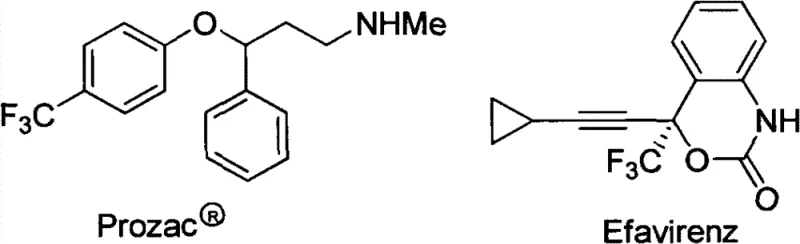 . However, the direct installation of this group often presents significant synthetic challenges due to the stability of the C-F bond and the harsh conditions typically required. Patent CN102153432B addresses these critical bottlenecks by disclosing a novel, highly efficient preparation method for alpha-trifluoromethyl-gamma-carbonyl esters. This technology represents a paradigm shift from traditional transition-metal catalysis to a pragmatic, metal-free cascade protocol that leverages widely available phosphorus ylides and trifluoropyruvates. For R&D directors and procurement specialists seeking reliable pharmaceutical intermediate supplier partnerships, this patent offers a robust pathway to access high-value fluorinated building blocks with superior cost-efficiency and scalability.
. However, the direct installation of this group often presents significant synthetic challenges due to the stability of the C-F bond and the harsh conditions typically required. Patent CN102153432B addresses these critical bottlenecks by disclosing a novel, highly efficient preparation method for alpha-trifluoromethyl-gamma-carbonyl esters. This technology represents a paradigm shift from traditional transition-metal catalysis to a pragmatic, metal-free cascade protocol that leverages widely available phosphorus ylides and trifluoropyruvates. For R&D directors and procurement specialists seeking reliable pharmaceutical intermediate supplier partnerships, this patent offers a robust pathway to access high-value fluorinated building blocks with superior cost-efficiency and scalability.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of complex fluorinated ketones and esters has relied heavily on transition-metal catalysis, specifically utilizing expensive and toxic heavy metals such as rhodium. A representative prior art method, depicted in Scheme 1 of the patent documentation, involves the reaction of silyl enol ethers with diazo compounds catalyzed by dirhodium tetraacetate. 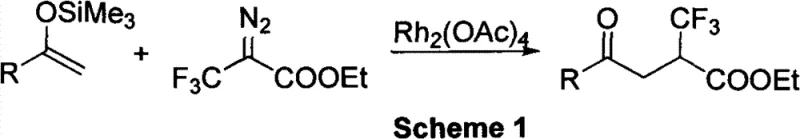 . This conventional approach suffers from multiple severe drawbacks that hinder its industrial applicability. Firstly, the requirement for rhodium catalysts introduces substantial raw material costs and necessitates rigorous downstream processing to ensure heavy metal residues are reduced to ppm levels acceptable for pharmaceutical use. Secondly, the diazo precursors used in these reactions are often unstable, potentially explosive, and require specialized handling protocols that increase operational risks. Furthermore, the preparation of the necessary silyl enol ether starting materials often demands strong bases and strictly anhydrous conditions, limiting the functional group tolerance and complicating the supply chain for diverse substrate libraries. These factors collectively result in a process that is economically burdensome and environmentally unsustainable for large-scale manufacturing.
. This conventional approach suffers from multiple severe drawbacks that hinder its industrial applicability. Firstly, the requirement for rhodium catalysts introduces substantial raw material costs and necessitates rigorous downstream processing to ensure heavy metal residues are reduced to ppm levels acceptable for pharmaceutical use. Secondly, the diazo precursors used in these reactions are often unstable, potentially explosive, and require specialized handling protocols that increase operational risks. Furthermore, the preparation of the necessary silyl enol ether starting materials often demands strong bases and strictly anhydrous conditions, limiting the functional group tolerance and complicating the supply chain for diverse substrate libraries. These factors collectively result in a process that is economically burdensome and environmentally unsustainable for large-scale manufacturing.
The Novel Approach
In stark contrast to the metal-dependent strategies of the past, the methodology disclosed in patent CN102153432B utilizes a clever tandem sequence involving a Wittig reaction followed by a hydrosilylation reduction. 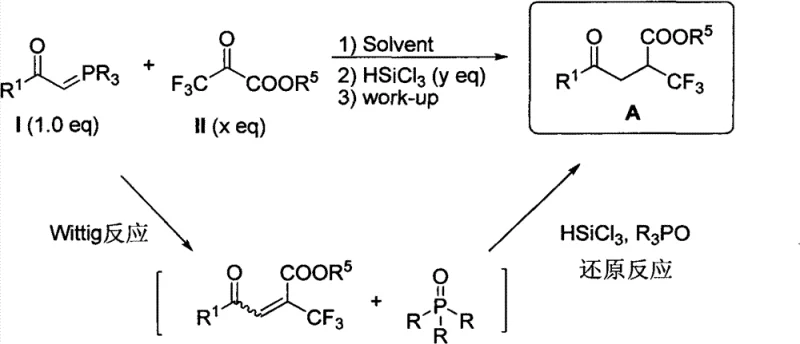 . This novel approach initiates with the reaction of a phosphorus ylide and ethyl trifluoropyruvate to form an alpha,beta-unsaturated intermediate, which is subsequently reduced in situ using trichlorosilane (HSiCl3). The brilliance of this design lies in its atom economy and operational simplicity; it avoids the isolation of unstable intermediates and eliminates the need for any transition metal catalysts entirely. By employing a cascade reaction strategy, the process effectively utilizes the phosphine oxide byproduct generated in the first step to potentially facilitate the second reduction step, thereby turning a waste product into a functional component of the reaction system. This results in a synthesis that is not only cheaper and safer but also exhibits remarkable compatibility with a wide array of functional groups, making it an ideal candidate for the cost reduction in pharmaceutical intermediate manufacturing.
. This novel approach initiates with the reaction of a phosphorus ylide and ethyl trifluoropyruvate to form an alpha,beta-unsaturated intermediate, which is subsequently reduced in situ using trichlorosilane (HSiCl3). The brilliance of this design lies in its atom economy and operational simplicity; it avoids the isolation of unstable intermediates and eliminates the need for any transition metal catalysts entirely. By employing a cascade reaction strategy, the process effectively utilizes the phosphine oxide byproduct generated in the first step to potentially facilitate the second reduction step, thereby turning a waste product into a functional component of the reaction system. This results in a synthesis that is not only cheaper and safer but also exhibits remarkable compatibility with a wide array of functional groups, making it an ideal candidate for the cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Wittig-Hydrosilylation Cascade
The mechanistic pathway of this transformation is a sophisticated interplay of nucleophilic addition and hydride transfer. Initially, the phosphorus ylide attacks the ketone carbonyl of the trifluoropyruvate ester, proceeding through a betaine or oxaphosphetane intermediate to yield the alpha,beta-unsaturated ester and triphenylphosphine oxide. Unlike traditional stepwise syntheses where this unsaturated species would be isolated, the patented process maintains the reaction mixture, allowing for the immediate introduction of trichlorosilane. The electron-deficient nature of the double bond, activated by both the carbonyl and the strongly electron-withdrawing trifluoromethyl group, makes it highly susceptible to conjugate reduction. The trichlorosilane acts as a hydride source, likely activated by the Lewis basicity of the phosphine oxide or the ester carbonyl, delivering a hydride to the beta-position. This generates a silyl enol ether transiently, which is subsequently hydrolyzed during the aqueous work-up to reveal the saturated gamma-keto ester. This mechanism ensures high regioselectivity for the 1,4-reduction over 1,2-reduction, preserving the ester functionality while installing the crucial trifluoromethyl group at the alpha-position relative to the ester.
From an impurity control perspective, this mechanism offers distinct advantages for ensuring high-purity OLED material or API intermediate production. The absence of transition metals removes a major class of difficult-to-remove impurities, simplifying the purification profile significantly. Furthermore, the mild reaction conditions, ranging from -40°C to 100°C depending on the substrate, prevent the thermal degradation of sensitive functional groups that might occur under harsher catalytic conditions. The patent data demonstrates that substrates containing nitro, chloro, methoxy, and even heteroaryl rings like thiophene and naphthalene are tolerated without side reactions such as dehalogenation or ring opening. This high chemoselectivity minimizes the formation of structurally related by-products, thereby enhancing the overall yield and reducing the burden on downstream chromatographic purification. The ability to tune the reaction by selecting appropriate solvents like dichloromethane, toluene, or THF further allows process chemists to optimize solubility and reaction kinetics for specific commercial scale-up of complex pharmaceutical intermediates.
How to Synthesize Alpha-Trifluoromethyl-Gamma-Carbonyl Ester Efficiently
The practical execution of this synthesis is designed for ease of operation in standard laboratory and pilot plant settings. The general procedure involves dissolving the phosphorus ylide in a suitable dry solvent under an inert nitrogen atmosphere to prevent oxidation or moisture interference. Once dissolved, the ethyl trifluoropyruvate is added at low temperatures, typically between -10°C and 0°C, to control the exotherm of the initial Wittig olefination. The mixture is then heated to a temperature between 0°C and 100°C, depending on the reactivity of the ylide, and stirred until the starting ylide is consumed as monitored by TLC. Upon completion of the first step, the reaction is cooled back to 0°C, and trichlorosilane is added carefully to effect the reduction. The detailed standardized synthesis steps see the guide below.
- Dissolve phosphorus ylide and solvent under nitrogen, then add ethyl trifluoropyruvate at low temperature (-10 to 0°C) and heat to 0-100°C to complete the Wittig reaction.
- Cool the mixture to 0°C and add trichlorosilane (HSiCl3) to initiate the conjugate reduction reaction, stirring until TLC indicates completion.
- Quench the reaction with an alcohol or salt solution, adsorb onto silica gel, and purify via column chromatography to isolate the target ester.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this technology translates into tangible strategic benefits regarding cost, reliability, and environmental compliance. The elimination of precious metal catalysts like rhodium removes a volatile cost component from the bill of materials, as the price of such metals can fluctuate wildly based on geopolitical factors. Moreover, the avoidance of heavy metals streamlines the quality control workflow, as there is no need for expensive ICP-MS testing or specialized scavenger resins to meet regulatory limits for metal residues in final drug substances. This directly contributes to substantial cost savings in the overall manufacturing budget.
- Cost Reduction in Manufacturing: The economic impact of this metal-free protocol is profound. By replacing expensive rhodium catalysts and unstable diazo reagents with commercially available phosphorus ylides and trifluoropyruvates, the raw material costs are drastically simplified. The cascade nature of the reaction means that two chemical transformations occur in a single vessel, which significantly reduces solvent consumption, energy usage for heating and cooling cycles, and labor hours associated with intermediate isolation and drying. This lean manufacturing approach ensures that the production of high-purity pharmaceutical intermediates remains economically viable even at smaller batch sizes, providing a competitive edge in pricing negotiations.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the use of commoditized reagents. Phosphorus ylides and ethyl trifluoropyruvate are produced by multiple global suppliers, reducing the risk of single-source dependency that often plagues specialized catalytic systems. The stability of these starting materials allows for long-term storage without significant degradation, enabling manufacturers to maintain strategic stockpiles against market disruptions. Furthermore, the robustness of the reaction conditions means that the process is less sensitive to minor variations in utility supplies or environmental controls, ensuring consistent batch-to-batch quality and reducing the lead time for high-purity pharmaceutical intermediates delivery to clients.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this process aligns perfectly with green chemistry principles. The absence of toxic heavy metals reduces the hazardous waste load, simplifying disposal and lowering treatment costs. The atom economy is improved by the tandem design, which minimizes waste generation per kilogram of product. Scalability is inherently supported by the mild thermal profile and the lack of hazardous gas evolution (unlike diazo decomposition), making the transition from gram-scale R&D to multi-ton commercial production smoother and safer. This facilitates the commercial scale-up of complex fluorinated intermediates while maintaining strict adherence to increasingly stringent global environmental regulations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and claims within patent CN102153432B, providing a factual basis for process evaluation. Understanding these details is crucial for technical teams assessing the feasibility of integrating this route into their existing manufacturing portfolios.
Q: What are the primary advantages of this metal-free synthesis over rhodium-catalyzed methods?
A: This method eliminates the need for expensive and toxic heavy metal catalysts like rhodium, significantly reducing raw material costs and simplifying the purification process by avoiding stringent heavy metal residue limits required for pharmaceutical APIs.
Q: How does the cascade reaction design improve atom economy?
A: By combining the Wittig olefination and the subsequent reduction in a single pot without isolating the intermediate unsaturated ester, the process minimizes solvent usage, reduces processing time, and cleverly utilizes the phosphine oxide byproduct to facilitate the reduction step.
Q: Is this synthesis compatible with sensitive functional groups?
A: Yes, the reaction conditions are extremely mild, operating between -40°C and 100°C without strong acids or bases, demonstrating high compatibility with various functional groups including nitro, chloro, methoxy, and heteroaryl moieties as evidenced by the broad substrate scope in the patent examples.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Alpha-Trifluoromethyl-Gamma-Carbonyl Ester Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced fluorination technologies play in the development of next-generation therapeutics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial reality is seamless. We are committed to delivering products with stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify the absence of metal residues and confirm structural integrity. Our capability to handle complex fluorinated chemistries positions us as a strategic partner for your long-term supply needs.
We invite you to leverage our technical expertise to optimize your supply chain. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific project requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our implementation of patent CN102153432B can enhance your production efficiency and reduce overall costs. Let us collaborate to bring your fluorinated drug candidates to market faster and more economically.