Scalable Synthesis of 3-Fluoroalkyl Chromone Derivatives for Advanced Pharmaceutical Applications
Introduction to Advanced Chromone Functionalization
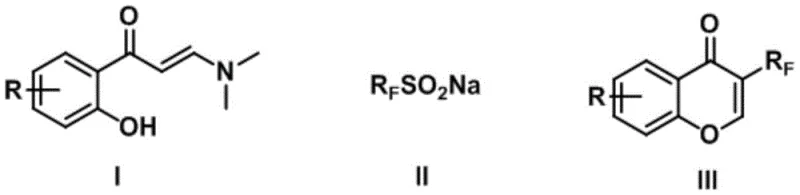
The pharmaceutical and agrochemical industries continuously seek efficient pathways to access fluorinated heterocycles, driven by the unique metabolic stability and bioactivity imparted by fluorine atoms. Patent CN114539197B discloses a groundbreaking synthesis method for 3-fluoroalkyl substituted chromone derivatives, addressing critical bottlenecks in existing manufacturing technologies. This innovation leverages visible-light photocatalysis or electrochemical oxidation to facilitate a tandem fluoroalkylation-cyclization sequence, transforming simple 2-hydroxyphenyl enaminones into valuable chromone scaffolds. By utilizing inexpensive organic photocatalysts such as Eosin Y or straightforward electrolytes, the process circumvents the reliance on precious metal catalysts that have historically inflated production costs. The methodology operates under remarkably mild conditions, typically between 10°C and 40°C, ensuring thermal safety and energy efficiency during operation. Furthermore, the reaction exhibits exceptional regioselectivity, exclusively functionalizing the C-3 position of the chromone ring, which is a pivotal site for biological activity in numerous drug candidates. This technical advancement represents a significant leap forward for reliable pharmaceutical intermediate suppliers aiming to optimize their synthetic portfolios.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of 3-fluoroalkyl chromones has been plagued by significant operational and economic challenges that hinder widespread industrial adoption. Traditional protocols often necessitate the use of costly transition metal photocatalysts, such as Iridium or Ruthenium polypyridyl complexes, which not only drive up raw material expenses but also introduce complex heavy metal removal steps during downstream processing. Additionally, many established routes rely on stoichiometric amounts of strong chemical oxidants like potassium persulfate or tert-butyl hydroperoxide, which pose safety hazards regarding storage and handling on a large scale. These oxidative conditions can sometimes lead to over-oxidation or degradation of sensitive functional groups on the aromatic ring, thereby compromising the overall yield and purity of the final product. The requirement for heating in some prior art methods further exacerbates energy consumption and limits the compatibility with thermally labile substrates. Consequently, procurement managers and process chemists have long struggled to find a balance between synthetic efficiency, cost containment, and environmental compliance when sourcing these critical intermediates.
The Novel Approach
The methodology outlined in the patent data introduces a paradigm shift by employing either visible-light photoredox catalysis or direct electrochemical oxidation to drive the transformation. In the photocatalytic variant, organic dyes like Eosin Y or Acr+-Mes·ClO4- serve as effective electron mediators, absorbing blue light to generate the necessary radical species without the need for rare earth metals. Alternatively, the electrochemical approach utilizes electricity as a traceless reagent, oxidizing the fluoroalkyl sulfinate directly at the anode surface to initiate the radical cascade. Both strategies operate at ambient or near-ambient temperatures, drastically reducing the thermal load on the reactor system. This dual-mode flexibility allows manufacturers to select the most economically viable option based on their existing infrastructure, whether it be a photo-reactor setup or an electrolytic cell. The avoidance of external chemical oxidants means that the only byproducts are benign salts, simplifying waste treatment and aligning with green chemistry principles. This novel approach effectively decouples high-yield synthesis from high operational costs, offering a robust solution for cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Photocatalytic and Electrochemical Cyclization
Understanding the mechanistic underpinnings of this transformation is crucial for R&D directors evaluating its feasibility for complex molecule synthesis. The reaction initiates with the single-electron oxidation of the sodium fluoroalkyl sulfinate species. In the photocatalytic cycle, the excited state of the organic photocatalyst acts as a potent oxidant, abstracting an electron from the sulfinate anion to generate a fluoroalkyl radical and sulfur dioxide. In the electrochemical mode, this oxidation occurs directly at the anode interface upon application of a specific current density. Once generated, the highly reactive fluoroalkyl radical adds regioselectively to the electron-deficient double bond of the enaminone substrate. This addition creates a new carbon-carbon bond and a transient carbon-centered radical intermediate adjacent to the carbonyl group. Subsequent intramolecular cyclization occurs via the attack of the phenolic oxygen onto the activated intermediate, closing the pyran ring. Finally, rearomatization through proton loss and oxidation restores the aromaticity of the system, delivering the stable 3-fluoroalkyl chromone product. This radical-polar crossover mechanism ensures high fidelity in bond formation, minimizing the formation of regioisomers that often complicate purification efforts in traditional electrophilic substitution reactions.
Impurity control is inherently built into this mechanism due to the mild nature of the radical generation and the specific reactivity of the enaminone double bond. Unlike harsh acidic or basic conditions that might promote hydrolysis of the enaminone or polymerization of the radical species, the neutral to slightly basic conditions maintained by the sulfinate salt preserve the integrity of the starting materials. The use of organic photocatalysts avoids the leaching of metal ions, which is a common source of contamination in API manufacturing that requires extensive scavenging procedures. Furthermore, the electrochemical method offers precise control over the oxidation potential by adjusting the current, preventing over-oxidation of the product or the substrate. This level of control results in cleaner reaction profiles, as evidenced by the high isolated yields reported across various substrates in the patent examples. For quality assurance teams, this translates to a more predictable impurity profile and reduced burden on analytical validation, facilitating faster regulatory approval for new drug applications containing these scaffolds.
How to Synthesize 3-Fluoroalkyl Chromone Derivatives Efficiently
Implementing this synthesis route requires careful attention to reaction parameters to maximize throughput and yield. The process begins by dissolving the 2-hydroxyphenyl enaminone and the sodium fluoroalkyl sulfinate in a polar aprotic solvent such as DMSO, acetonitrile, or acetone. For the photocatalytic variation, a catalytic amount of Eosin Y is added, and the mixture is irradiated with a blue LED light source while stirring at room temperature. Alternatively, for the electrochemical variation, an undivided cell equipped with a graphite anode and a platinum or iron cathode is utilized, with a supporting electrolyte like tetrabutylammonium tetrafluoroborate added to ensure conductivity. The reaction progress is monitored until the starting material is consumed, typically within 8 to 24 hours depending on the specific substrate and fluoroalkyl chain length. Upon completion, the reaction mixture undergoes a standard aqueous workup involving water washing and extraction with ethyl acetate. The crude product is then dried and purified via silica gel column chromatography to afford the pure 3-fluoroalkyl chromone derivative. Detailed standardized synthesis steps are provided in the guide below.
- Dissolve the 2-hydroxyphenyl enaminone substrate and sodium fluoroalkyl sulfinate in a suitable solvent such as DMSO or acetonitrile.
- Add an organic photocatalyst like Eosin Y or an electrolyte like tetrabutylammonium tetrafluoroborate depending on the chosen activation mode.
- Irradiate with blue light for photocatalysis or apply constant current for electrochemistry, then purify the resulting chromone derivative via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic sourcing perspective, this patented technology offers compelling advantages that directly impact the bottom line and supply chain resilience. The elimination of expensive noble metal catalysts represents a direct reduction in raw material costs, which is particularly significant given the volatility of prices for Iridium and Ruthenium compounds. Moreover, the absence of strong chemical oxidants reduces the hazard classification of the process, potentially lowering insurance premiums and safety compliance costs associated with storing hazardous reagents. The mild reaction temperatures allow for the use of standard glass-lined or stainless steel reactors without the need for specialized high-pressure or high-temperature equipment, thereby increasing the available capacity within existing manufacturing facilities. This accessibility facilitates rapid scale-up from laboratory benchtop to commercial production volumes without significant capital expenditure on new infrastructure. Additionally, the broad substrate tolerance means that a single platform technology can be adapted to produce a wide library of fluorinated chromones, enhancing supply chain flexibility and reducing the need for multiple specialized synthesis lines.
- Cost Reduction in Manufacturing: The substitution of precious metal photocatalysts with abundant organic dyes or electricity drastically lowers the cost of goods sold (COGS). By removing the need for post-reaction metal scavenging resins, the downstream processing becomes simpler and less expensive. The atom economy is improved by avoiding stoichiometric oxidants, meaning less waste is generated per kilogram of product. These factors combine to create a leaner manufacturing process that is highly competitive in price-sensitive markets. The qualitative shift from metal-based to organic or electro-based catalysis ensures long-term cost stability不受 fluctuating metal market prices.
- Enhanced Supply Chain Reliability: The reagents required for this synthesis, including sodium fluoroalkyl sulfinates and organic dyes, are commercially available from multiple global vendors, reducing the risk of single-source supply disruptions. The robustness of the reaction conditions means that production is less susceptible to delays caused by equipment failure or strict environmental controls needed for hazardous oxidants. This reliability ensures consistent delivery schedules for downstream customers, which is critical for maintaining continuous API production lines. The ability to run reactions at ambient temperature also reduces dependence on complex utility systems for heating or cooling, further stabilizing the manufacturing timeline against utility fluctuations.
- Scalability and Environmental Compliance: The green chemistry credentials of this method align perfectly with increasingly stringent environmental regulations globally. The reduction in hazardous waste streams simplifies effluent treatment and lowers disposal costs. The electrochemical option, in particular, offers a 'reagent-free' oxidation pathway that minimizes the chemical footprint of the process. Scalability is enhanced because both photo-flow reactors and electrochemical cells are well-established technologies for continuous manufacturing, allowing for seamless transition from batch to flow processing. This future-proofs the supply chain against regulatory changes and supports sustainability goals that are becoming mandatory for major pharmaceutical buyers.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and claims within the patent documentation to ensure accuracy and relevance for technical decision-makers. Understanding these details helps in assessing the fit of this technology within your specific project requirements and quality standards. We encourage further discussion with our technical team to explore custom adaptations for your specific molecular targets.
Q: What are the primary advantages of this synthesis method over traditional routes?
A: This method avoids the use of expensive transition metal photocatalysts like Iridium or Ruthenium complexes and eliminates the need for harsh chemical oxidants, resulting in a greener and more cost-effective process.
Q: Can this protocol accommodate diverse fluoroalkyl groups?
A: Yes, the protocol demonstrates excellent substrate scope, successfully incorporating trifluoromethyl, perfluorobutyl, perfluorohexyl, and perfluorooctyl groups with high regioselectivity.
Q: Is this method suitable for large-scale industrial production?
A: The mild reaction conditions, typically ranging from 10°C to 40°C, and the use of readily available organic dyes or electricity make this process highly scalable and safe for commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Fluoroalkyl Chromone Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this photocatalytic and electrochemical synthesis route for producing high-value fluorinated intermediates. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from development to market. Our facility is equipped with state-of-the-art photo-reactors and electrochemical synthesis units capable of handling the specific requirements of this patent technology. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of 3-fluoroalkyl chromone meets the highest international standards for pharmaceutical use. Our commitment to quality ensures that the impurity profiles are tightly controlled, facilitating your regulatory filings.
We invite you to collaborate with us to leverage this innovative chemistry for your next drug development program. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and route feasibility assessments to demonstrate how this method can optimize your supply chain. Contact us today to discuss how we can support your need for high-purity pharmaceutical intermediates with reliable, scalable, and cost-effective manufacturing solutions.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →