Revolutionizing Fluorinated Building Blocks: Scalable Synthesis of Monofluoro Iodoacetone Compounds
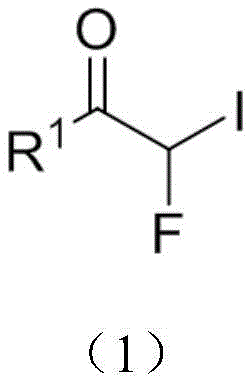
The landscape of fluorinated organic synthesis is undergoing a significant transformation, driven by the critical need for stable, versatile building blocks in modern drug discovery. Patent CN113929569A introduces a groundbreaking methodology for the preparation of monofluoro iodoacetone compounds, a class of molecules that serves as a pivotal universal building block for complex organic synthesis. These compounds, characterized by the presence of both a fluorine and an iodine atom on the alpha-carbon relative to a carbonyl group, offer unique reactivity profiles that are essential for introducing monofluoroalkyl groups into pharmaceutical scaffolds. The patent details a robust synthetic route that overcomes the historical limitations of instability and harsh conditions associated with earlier generation fluorinating reagents. By leveraging a specific combination of lithium iodide and selective halogenating agents, this technology enables the efficient production of high-purity intermediates suitable for large-scale manufacturing.
For R&D directors and process chemists, the structural versatility of these compounds cannot be overstated. The general formula allows for a wide array of R1 substituents, ranging from simple phenyl groups to complex heterocycles and condensed ring systems. This flexibility means that a single synthetic platform can generate a diverse library of fluorinated ketones, accelerating the lead optimization phase in medicinal chemistry programs. Furthermore, the ability to introduce a monofluoro methyl group with high precision addresses a key challenge in metabolic stability enhancement, making these intermediates highly valuable assets in the development of next-generation active pharmaceutical ingredients (APIs).
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the introduction of monofluoroalkyl groups into organic molecules has been fraught with synthetic challenges. Conventional reagents such as BrCFHCOOEt, (EtO)2P(O)CFHBr, or PhSO2CHFI often suffer from significant drawbacks that hinder their widespread adoption in industrial settings. Many of these traditional reagents are thermally unstable, requiring stringent storage conditions and careful handling to prevent decomposition before they can even react. Moreover, the reaction conditions required to activate these reagents are frequently harsh, involving extreme temperatures or strong bases that are incompatible with sensitive functional groups commonly found in advanced drug intermediates. This lack of functional group tolerance often necessitates additional protection and deprotection steps, drastically increasing the step count, reducing overall yield, and inflating the cost of goods sold (COGS). Additionally, the purification of products derived from these older methods can be cumbersome, often leaving behind difficult-to-remove impurities that compromise the quality of the final API.
The Novel Approach
In stark contrast, the methodology disclosed in CN113929569A represents a paradigm shift towards efficiency and practicality. The novel approach utilizes a gem-diol precursor, specifically a tetrafluoro-dihydroxy ketone derivative, which reacts with lithium iodide and a halogenating agent such as Selectfluor, NIS, or NBS. This reaction proceeds under remarkably mild conditions, typically initiating in an ice bath and completing at temperatures ranging from -20°C to 40°C. The reaction time is significantly shortened, often requiring only 10 to 40 minutes in the ice bath followed by a brief period at room temperature. This dramatic reduction in thermal stress not only preserves the integrity of sensitive substrates but also translates to substantial energy savings in a commercial plant setting. The use of commercially available and stable reagents like lithium iodide and triethylamine further simplifies the supply chain logistics, ensuring that production can be scaled up without the bottlenecks associated with exotic or hazardous reagents.

Mechanistic Insights into Lithium Iodide Mediated Halogen Exchange
The success of this synthetic strategy lies in the intricate interplay between the iodide source and the oxidizing halogenating agent. Mechanistically, the lithium iodide serves as a source of nucleophilic iodide ions, which interact with the electrophilic fluorine or halogen species generated from reagents like Selectfluor, NIS, or NBS. This interaction facilitates a halogen exchange process that ultimately generates reactive iodine species in situ. A critical component of this mechanism is the role of triethylamine, which is added after the initial mixing. Triethylamine acts not merely as a base to neutralize acidic byproducts but plays a sophisticated role in polarizing the iodine molecules generated during the reaction. By inducing a dipole in the I-I bond, triethylamine creates a partial positive charge on one iodine atom, making it highly susceptible to nucleophilic attack by the enolate form of the ketone precursor. This polarization effect accelerates the reaction kinetics, allowing the transformation to proceed rapidly even at near-ambient temperatures. The result is a clean conversion to the desired alpha-fluoro-alpha-iodo ketone with minimal formation of side products such as non-iodinated monofluoro ketones.
From an impurity control perspective, this mechanism offers distinct advantages. The specificity of the enolate attack on the polarized iodine species ensures high regioselectivity, minimizing the risk of poly-halogenation or rearrangement byproducts that often plague radical fluorination methods. Furthermore, the byproducts of this reaction, primarily lithium salts and succinimide derivatives (if NIS/NBS are used), are generally water-soluble or easily removed during the aqueous workup phase described in the patent examples. This inherent cleanliness of the reaction profile simplifies downstream processing, reducing the burden on purification columns and crystallization steps. For process chemists, understanding this mechanism is key to optimizing the molar ratios; the patent suggests a molar ratio of precursor to halogenating agent between 1:2 and 1:10, and precursor to lithium iodide between 1:2 and 1:8, providing a wide operational window that enhances process robustness against minor variations in feedstock quality.
How to Synthesize Monofluoro Iodoacetone Efficiently
The practical implementation of this synthesis involves a straightforward sequence of mixing, cooling, and basification that is amenable to standard reactor setups. The process begins with the dissolution of the tetrafluoro-dihydroxy ketone precursor in a suitable organic solvent such as tetrahydrofuran (THF), 1,4-dioxane, or acetonitrile. Following the addition of lithium iodide and the chosen halogenating agent, the mixture is stirred under cooling to control the exotherm and ensure selectivity. The subsequent addition of triethylamine triggers the final cyclization/substitution event, driving the reaction to completion within a few hours. Detailed standard operating procedures regarding exact stoichiometry, addition rates, and specific workup protocols are critical for reproducibility and safety.
- Mix the gem-diol precursor compound with lithium iodide and a halogenating agent (Selectfluor, NIS, or NBS) in an organic solvent like THF.
- Stir the reaction mixture in an ice-water bath for 10 to 40 minutes to initiate the halogen exchange.
- Add triethylamine slowly, allow the reaction to proceed at -20°C to 40°C (preferably 25°C) for 0.5 to 3 hours, then purify via extraction and chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this technology offers compelling economic and logistical benefits that extend beyond simple yield improvements. The shift towards milder reaction conditions directly correlates with reduced operational expenditures (OPEX) in a manufacturing environment. By eliminating the need for deep cryogenic cooling (often below -78°C) or prolonged heating cycles, the energy consumption per kilogram of product is significantly lowered. This energy efficiency is a crucial factor in maintaining competitive pricing in the global market for fine chemical intermediates. Moreover, the reagents employed—lithium iodide, triethylamine, and common halogenating agents—are commodity chemicals with stable supply chains. This reduces the risk of production delays caused by the scarcity of specialized reagents, ensuring a consistent and reliable flow of materials for continuous manufacturing campaigns.
- Cost Reduction in Manufacturing: The streamlined nature of this synthesis eliminates several cost drivers associated with traditional fluorination methods. The high reaction efficiency and short reaction times mean that reactor occupancy is minimized, allowing for higher throughput in existing facilities without the need for capital-intensive expansion. Additionally, the simplified workup procedure, which typically involves standard aqueous extraction and silica gel chromatography, reduces the consumption of expensive solvents and stationary phases. The avoidance of transition metal catalysts in the primary synthesis step further lowers costs by removing the need for expensive metal scavengers and rigorous heavy metal testing, which are mandatory for pharmaceutical grade materials. These cumulative factors contribute to a substantially lower cost of goods, enabling more aggressive pricing strategies while maintaining healthy margins.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the versatility of the starting materials. The R1 group on the precursor can be easily varied, meaning that a single manufacturing line can be adapted to produce a wide range of different monofluoro iodoacetone derivatives simply by changing the starting ketone. This flexibility allows manufacturers to respond rapidly to shifting market demands or specific client requests for custom analogs. Furthermore, the stability of the intermediates and the robustness of the reaction conditions reduce the likelihood of batch failures due to environmental fluctuations or minor procedural deviations. This reliability translates into shorter lead times for customers, as manufacturers can confidently commit to delivery schedules without the buffer time usually required for troubleshooting unpredictable chemistries.
- Scalability and Environmental Compliance: Scaling this process from gram to ton scale is facilitated by the absence of hazardous gases or highly unstable intermediates. The reaction generates minimal hazardous waste, and the solvents used are standard organic liquids that can be readily recovered and recycled through distillation. This aligns with increasingly stringent environmental regulations and corporate sustainability goals. The ability to run the reaction at near-ambient temperatures also reduces the carbon footprint of the manufacturing process. For supply chain planners, this means fewer regulatory hurdles and a smoother path to commercial approval for new drug applications that utilize these intermediates, as the environmental, health, and safety (EHS) profile of the process is favorable compared to older, more toxic fluorination technologies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these fluorinated building blocks. These insights are derived directly from the experimental data and technical disclosures within the patent literature, providing a factual basis for decision-making.
Q: What are the primary advantages of this synthesis method over traditional monofluoroalkylation reagents?
A: Unlike traditional reagents which may be unstable or require harsh conditions, this method utilizes mild reaction temperatures (-20°C to 40°C) and short reaction times, resulting in higher yields and easier purification.
Q: What is the scope of substituents compatible with this reaction?
A: The process is highly versatile, accommodating R1 groups that include phenyl, condensed rings, substituted phenyls (with F, Cl, Br, CF3, OCF3, etc.), alkyl chains, and heterocycles.
Q: How does the use of triethylamine impact the reaction mechanism?
A: Triethylamine plays a critical role in polarizing iodine molecules generated in situ, facilitating the formation of electrophilic iodine species that are attacked by enolates to form the final product.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Monofluoro Iodoacetone Supplier
As the demand for fluorinated pharmaceutical intermediates continues to surge, partnering with a manufacturer that possesses deep technical expertise and scalable infrastructure is paramount. NINGBO INNO PHARMCHEM stands at the forefront of this sector, leveraging advanced synthetic methodologies like the one described in CN113929569A to deliver high-purity monofluoro iodoacetone compounds. Our team of expert chemists has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements whether you are in the pre-clinical stage or full-scale commercial manufacturing. We adhere to stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch meets the exacting standards required for GMP environments.
We invite you to collaborate with us to optimize your supply chain for fluorinated intermediates. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific project needs, demonstrating how our efficient synthesis routes can reduce your overall material costs. Contact us today to request specific COA data for our catalog items or to discuss route feasibility assessments for your custom targets. Let us be your strategic partner in bringing innovative fluorinated medicines to market faster and more economically.