Advanced Direct Alkylation Strategy for High-Purity 2'-O-(2-Methoxyethyl) Guanosine Manufacturing
The rapid expansion of the gene therapy market, particularly in the realm of antisense oligonucleotides (ASOs), has placed immense pressure on the supply chain for modified nucleoside building blocks. Among these, 2'-O-(2-methoxyethyl) guanosine stands out as a critical monomer for enhancing the binding affinity and nuclease resistance of therapeutic oligonucleotides. A recent technological breakthrough documented in patent CN114835765B introduces a highly efficient, one-step synthesis process that fundamentally alters the production landscape for this valuable intermediate. By shifting away from multi-step protection-deprotection sequences and enzymatic conversions, this innovation offers a direct alkylation pathway starting from readily available guanosine. For pharmaceutical manufacturers and CDMOs, this represents a pivotal shift towards more robust, cost-effective, and scalable manufacturing protocols that align with the rigorous demands of modern small nucleic acid drug development.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2'-O-(2-methoxyethyl) guanosine has been plagued by complex, multi-stage routes that introduce significant bottlenecks in both cost and operational safety. Early methodologies, such as those disclosed in US20080234475 A1, relied on the use of tris(2-methoxyethyl) borate as an alkylating agent, necessitating high-temperature reactions that are extremely sensitive to water content. Furthermore, these legacy processes often required a subsequent desulfurization step using Raney nickel, a pyrophoric catalyst that poses severe safety hazards and creates substantial environmental disposal challenges. 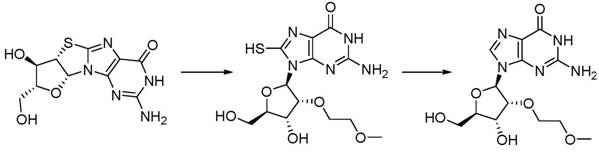 Other approaches attempted to solve selectivity issues by employing bulky protecting groups like MDPSCl2 to mask the 3' and 5' hydroxyls, as seen in WO 2003087053A 2. However, these protecting groups are not only prohibitively expensive for large-scale production but also require harsh fluoride reagents for removal, complicating the purification profile and introducing difficult-to-remove impurities into the final API intermediate.
Other approaches attempted to solve selectivity issues by employing bulky protecting groups like MDPSCl2 to mask the 3' and 5' hydroxyls, as seen in WO 2003087053A 2. However, these protecting groups are not only prohibitively expensive for large-scale production but also require harsh fluoride reagents for removal, complicating the purification profile and introducing difficult-to-remove impurities into the final API intermediate.
The Novel Approach
In stark contrast to these convoluted pathways, the process described in CN114835765B achieves the transformation through a direct, chemoselective alkylation of the 2'-hydroxyl group on the ribose sugar. This novel approach eliminates the need for temporary protecting groups on the sugar moiety and avoids the use of enzymatic biocatalysts entirely. By carefully tuning the reaction environment—specifically the choice of base, solvent, and optional iodine catalyst—the method successfully directs the alkylation to the oxygen atom rather than the nitrogen atoms on the guanine base. 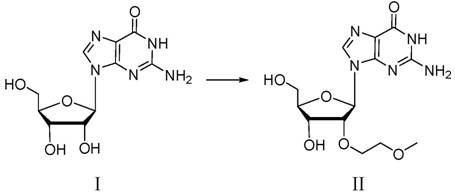 This simplification reduces the total number of unit operations, minimizes solvent consumption, and drastically shortens the overall production timeline. The ability to proceed directly from guanosine to the target molecule without intermediate isolation steps represents a significant leap forward in process chemistry efficiency, making it ideally suited for the high-volume demands of the growing oligonucleotide therapeutics market.
This simplification reduces the total number of unit operations, minimizes solvent consumption, and drastically shortens the overall production timeline. The ability to proceed directly from guanosine to the target molecule without intermediate isolation steps represents a significant leap forward in process chemistry efficiency, making it ideally suited for the high-volume demands of the growing oligonucleotide therapeutics market.
Mechanistic Insights into Base-Catalyzed Direct Alkylation
The core of this synthetic advancement lies in the precise control of nucleophilicity within the guanosine molecule. Under standard conditions, the nitrogen atoms of the purine ring are competitive nucleophiles, often leading to unwanted N-alkylated byproducts, specifically at the O6 position which can tautomerize. The patented process overcomes this by utilizing strong, non-nucleophilic bases such as sodium bis(trimethylsilyl)amide (NaHMDS) or inorganic bases like potassium hydroxide in polar aprotic solvents like DMSO or DMF. These conditions facilitate the selective deprotonation of the 2'-hydroxyl group, generating a reactive alkoxide species that attacks the alkylating agent (e.g., 2-bromoethyl methyl ether) with high specificity. The inclusion of catalytic amounts of iodide salts, such as potassium iodide or tetrabutylammonium iodide, further enhances the reaction kinetics via a Finkelstein-type mechanism when using chloro- or bromo-alkylating agents, ensuring complete conversion even at moderate temperatures ranging from 10°C to 80°C.
From an impurity control perspective, this mechanism is superior because it avoids the formation of complex protected intermediates that often carry over impurities into the final step. The reaction mixture is designed to be heterogeneous or easily manageable, allowing for a straightforward workup procedure. Upon completion, the product can be precipitated directly from the reaction solution by adding a poor solvent such as acetonitrile or ethyl acetate. This crystallization-induced isolation not only serves as a primary purification step to remove unreacted starting materials and inorganic salts but also ensures that the final crude product possesses high purity levels, often exceeding 97% by HPLC before final chromatographic polishing. This robustness in impurity profiling is critical for meeting the stringent quality standards required for GMP manufacturing of nucleic acid drugs.
How to Synthesize 2'-O-(2-Methoxyethyl) Guanosine Efficiently
The implementation of this synthesis route requires careful attention to reagent stoichiometry and temperature control to maximize yield and minimize side reactions. The process is versatile, accommodating various alkylating agents including 2-chloroethyl methyl ether, 2-bromoethyl methyl ether, and sulfonate esters, providing flexibility based on raw material availability and cost. The reaction typically proceeds by dissolving guanosine in a solvent like DMSO, adding the base and catalyst, and then introducing the alkylating agent. Following the reaction period, which can range from a few hours to overnight depending on the specific reagents used, the product is isolated via filtration or centrifugation after anti-solvent addition.
- Dissolve guanosine in a polar aprotic solvent such as DMSO or DMF under inert atmosphere.
- Add a suitable base (e.g., KOH, NaHMDS) and an iodine catalyst if using chloro/bromo alkylating agents.
- React with 2-methoxyethyl halide or sulfonate at controlled temperatures (10-80°C) followed by precipitation purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this direct alkylation technology translates into tangible strategic benefits regarding cost stability and supply security. The most immediate impact is the drastic reduction in raw material costs associated with the elimination of exotic protecting groups and enzymes. Traditional routes relying on MDPSCl2 or adenosine deaminase create dependencies on niche suppliers with limited capacity, whereas this new method utilizes commodity chemicals like guanosine, potassium hydroxide, and simple alkyl halides that are available from multiple global sources. This diversification of the supply base significantly mitigates the risk of raw material shortages and price volatility, ensuring a more predictable cost structure for long-term production contracts.
- Cost Reduction in Manufacturing: The economic advantage of this process is driven primarily by the simplification of the synthetic route. By collapsing what was previously a three or four-step sequence into a single pot reaction, manufacturers save substantially on labor, energy, and solvent usage. The removal of the desulfurization step eliminates the need for specialized filtration equipment and hazardous waste handling associated with heavy metal catalysts. Furthermore, the avoidance of expensive silyl protecting groups and fluoride deprotection reagents removes some of the highest cost drivers from the bill of materials. These cumulative efficiencies result in a significantly lower cost of goods sold (COGS), allowing for more competitive pricing in the tendering process for oligonucleotide APIs.
- Enhanced Supply Chain Reliability: Supply continuity is often compromised by complex processes that have multiple failure points. This streamlined methodology reduces the number of intermediate quality control checkpoints and minimizes the potential for batch failures due to cumulative yield losses across multiple steps. The use of robust, non-sensitive reagents means that the process is less susceptible to variations in raw material quality or minor fluctuations in environmental conditions. Consequently, production lead times can be shortened, and the reliability of delivery schedules is improved, which is essential for maintaining the inventory levels required by just-in-time pharmaceutical manufacturing models.
- Scalability and Environmental Compliance: From an EHS (Environment, Health, and Safety) perspective, this process offers a cleaner profile that facilitates easier regulatory approval and plant permitting. The absence of pyrophoric Raney nickel and toxic fluoride sources simplifies waste stream management and reduces the burden on effluent treatment plants. The reaction conditions are mild enough to be performed in standard glass-lined or stainless steel reactors without requiring exotic metallurgy, making the technology highly transferable between different manufacturing sites. This inherent scalability ensures that production can be ramped up from pilot kilogram quantities to multi-ton commercial volumes without the need for fundamental process re-engineering.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. They are derived from the specific advantages outlined in the patent documentation and are intended to clarify the operational feasibility for potential partners.
Q: How does this process avoid N-alkylation byproducts?
A: The process utilizes specific base strengths and solvent systems that favor the nucleophilicity of the 2'-hydroxyl group over the nitrogen atoms on the guanine base, minimizing O6-alkylation impurities.
Q: What are the scalability advantages of this method?
A: By eliminating expensive protecting groups like MDPSCl2 and hazardous steps like Raney nickel desulfurization, the process reduces equipment complexity and safety risks, facilitating easier industrial scale-up.
Q: Is enzymatic conversion required?
A: No, unlike prior art methods that relied on adenosine deaminase enzymes, this chemical synthesis route is purely synthetic, removing the need for specialized biocatalytic conditions and buffers.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2'-O-(2-Methoxyethyl) Guanosine Supplier
As the demand for antisense therapies continues to surge, securing a dependable source of high-quality nucleoside intermediates is paramount for drug developers. NINGBO INNO PHARMCHEM leverages deep expertise in nucleoside chemistry to offer this advanced synthesis route, ensuring that clients receive material that meets the highest standards of purity and consistency. Our facilities are equipped to handle the specific solvent systems and reaction conditions required for this process, with extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of 2'-O-(2-methoxyethyl) guanosine is fully characterized and compliant with industry expectations.
We invite pharmaceutical companies and research institutions to collaborate with us to optimize their supply chains for next-generation genetic medicines. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact us directly to request specific COA data and route feasibility assessments, allowing us to demonstrate how our optimized manufacturing capabilities can support your clinical and commercial timelines effectively.