Advanced Synthesis of Chlorpheniramine Maleate Impurity for Precision Quality Control
Advanced Synthesis of Chlorpheniramine Maleate Impurity for Precision Quality Control
In the highly regulated landscape of pharmaceutical manufacturing, the ability to identify and quantify trace impurities is paramount for ensuring patient safety and regulatory compliance. Patent CN111100067A introduces a significant breakthrough in this domain by disclosing a novel chlorpheniramine maleate impurity and, crucially, a robust preparation process for its synthesis. This specific impurity is structurally analogous to the active pharmaceutical ingredient (API), chlorpheniramine maleate, which historically has presented substantial challenges in separation and identification during quality control assays. By providing a dedicated synthetic route to generate this compound in high purity, the patent offers the industry a vital tool for establishing accurate reference standards. This development not only enhances the precision of high-performance liquid chromatography (HPLC) detection methods but also streamlines the quality assurance protocols for antihistamine formulations globally.
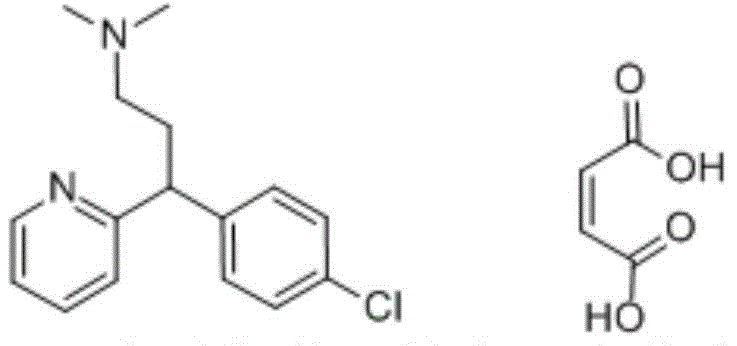
The structural proximity of this impurity to the parent drug means that without a pure reference standard, distinguishing between the API and the contaminant becomes analytically ambiguous. The invention addresses this by defining a clear chemical pathway that transforms accessible precursors into the target impurity through a logical sequence of organic transformations. For R&D directors and quality control managers, access to such well-characterized reference substances is indispensable for validating analytical methods and setting appropriate acceptance criteria for finished drug products. The ability to synthesize this molecule reliably removes the bottleneck of relying on unpredictable isolation from crude reaction mixtures, thereby stabilizing the supply chain for critical quality control materials.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the acquisition of specific process-related impurities for pharmaceutical reference standards has been fraught with inefficiency and inconsistency. In many legacy workflows, manufacturers attempt to isolate these trace contaminants directly from the bulk synthesis of the API or through non-specific degradation studies. This approach is inherently flawed because the concentration of such impurities in the crude mixture is often negligible, making isolation via preparative chromatography labor-intensive, costly, and low-yielding. Furthermore, the structural similarity between chlorpheniramine maleate and its impurities means that separation factors are often poor, requiring extensive optimization of mobile phases and stationary phases to achieve even marginal purity. This reliance on isolation creates a supply chain vulnerability where the availability of the reference standard is tied to the production volume and variability of the API itself, leading to potential delays in method validation and batch release testing.
The Novel Approach
The methodology outlined in CN111100067A represents a paradigm shift from isolation to dedicated synthesis. Instead of scavenging for trace amounts, the patent describes a constructive three-step synthetic route designed specifically to build the impurity molecule from simpler, commercially available building blocks. This approach leverages classic organic reactions—alkylation, hydrolysis, and condensation—that are well-understood and easily scalable. By decoupling the production of the reference standard from the main API manufacturing line, companies can produce large batches of the impurity independently. This ensures a steady inventory of high-purity material (exemplified in the patent at 99% purity) that can be used repeatedly for calibration and system suitability testing. The strategic advantage lies in the reproducibility of the synthesis; unlike isolation, which varies with every API batch, a dedicated synthetic process can be standardized, validated, and scaled to meet the growing demands of global regulatory bodies for rigorous impurity profiling.
Mechanistic Insights into the Three-Step Synthetic Route
The core of this innovation lies in a concise three-step reaction sequence that efficiently constructs the target molecular architecture. The process begins with the alkylation of a nitrile-containing precursor (Compound 1), which serves as the foundational scaffold. In this initial step, the acidic proton alpha to the nitrile group is deprotonated by a strong base, specifically sodium ethoxide, generating a nucleophilic carbanion. This reactive species then attacks bromoacetaldehyde diethyl acetal in an SN2 fashion, facilitated by the phase transfer catalyst tetrabutylammonium bromide in a toluene solvent system. This alkylation installs the necessary two-carbon chain bearing the protected aldehyde functionality, setting the stage for subsequent functional group manipulations. The use of the acetal protecting group is a critical mechanistic choice, as it prevents premature side reactions of the aldehyde during the harsh basic conditions of the alkylation step.
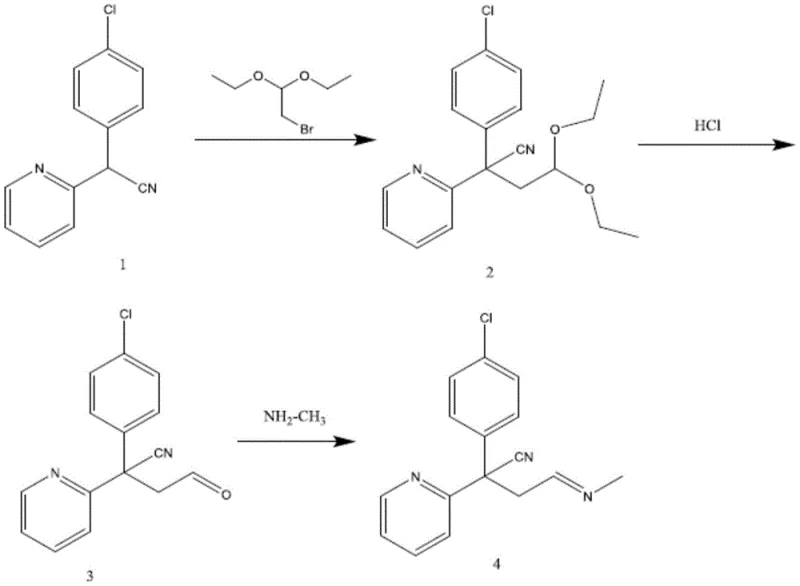
Following the successful construction of the carbon skeleton, the second step involves the deprotection of the acetal to reveal the reactive aldehyde moiety. This is achieved through acidic hydrolysis using hydrochloric acid in an acetone solvent under reflux conditions. The acid catalyzes the cleavage of the ether bonds in the acetal, releasing ethanol and generating the free aldehyde (Compound 3). This intermediate is pivotal, as the aldehyde group is the electrophilic center required for the final bond formation. The final step is a condensation reaction between this newly formed aldehyde and methylamine. Conducted in tetrahydrofuran (THF) at temperatures ranging from 0°C to room temperature, this reaction forms an imine (Schiff base) linkage. The presence of a drying agent, such as magnesium sulfate, drives the equilibrium forward by sequestering the water byproduct, ensuring high conversion to the final chlorpheniramine impurity. This mechanistic pathway is elegant in its simplicity, avoiding exotic reagents while delivering a molecule that mimics the structural nuances of the natural process impurity.
From an impurity control perspective, understanding this mechanism is vital for identifying potential byproducts. For instance, incomplete hydrolysis in step two could lead to residual acetal impurities, while over-alkylation in step one is a risk if stoichiometry is not tightly controlled. However, the patent details specific purification protocols, such as silica gel column chromatography with a dichloromethane and methanol eluent system, which effectively remove these side products. The result is a reference substance of exceptional purity, capable of serving as a definitive marker in HPLC assays. This level of chemical fidelity ensures that when the impurity is detected in a commercial batch of chlorpheniramine maleate, it can be quantified with absolute confidence, safeguarding the integrity of the final medicinal product.
How to Synthesize Chlorpheniramine Impurity Efficiently
The synthesis of this critical reference standard is designed to be operationally straightforward, utilizing common laboratory equipment and reagents to ensure accessibility for quality control laboratories and pilot plants alike. The process flow moves logically from the construction of the carbon backbone to the unveiling of the reactive functional groups, culminating in the formation of the target imine structure. Each step has been optimized in the patent examples to balance reaction rate with selectivity, minimizing the formation of difficult-to-remove byproducts. For technical teams looking to implement this route, the detailed procedural guidelines provided in the patent serve as a robust starting point for method transfer and scale-up activities. The following section outlines the standardized operational framework derived from the patent disclosure.
- Alkylation of the nitrile precursor with bromoacetaldehyde diethyl acetal using sodium ethoxide and tetrabutylammonium bromide in toluene.
- Acidic hydrolysis of the acetal intermediate using hydrochloric acid in acetone under reflux conditions to generate the aldehyde.
- Condensation of the aldehyde intermediate with methylamine in tetrahydrofuran (THF) using magnesium sulfate as a drying agent to form the final imine impurity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this synthetic route offers tangible benefits that extend beyond mere technical feasibility. The primary advantage is the stabilization of the supply chain for critical reference materials. By shifting from an isolation-dependent model to a synthesis-driven model, organizations eliminate the volatility associated with extracting trace impurities from variable API batches. This transition ensures that quality control laboratories have uninterrupted access to the standards they need to release batches of chlorpheniramine maleate, preventing costly delays in product distribution. Furthermore, the raw materials required for this synthesis, such as p-chlorobenzonitrile and 2-chloropyridine derivatives, are commodity chemicals with established global supply chains, reducing the risk of raw material shortages.
- Cost Reduction in Manufacturing: The economic impact of this process is driven by its efficiency and the elimination of wasteful isolation procedures. Traditional isolation of trace impurities often consumes vast quantities of solvents and chromatography media to recover milligram amounts of material, resulting in an exorbitant cost per gram. In contrast, this dedicated synthesis builds the molecule from scratch with high atom economy in the key steps. The use of standard solvents like toluene, acetone, and THF, which are inexpensive and readily recyclable, further drives down the operational expenditure. Additionally, the high purity achieved (up to 99% in exemplified runs) reduces the need for repetitive purification cycles, saving both time and resources. This streamlined approach translates into significant cost savings for the production of reference standards, allowing budget reallocation to other critical R&D initiatives.
- Enhanced Supply Chain Reliability: Reliability in the supply of reference standards is a cornerstone of consistent pharmaceutical manufacturing. This synthetic route decouples the availability of the impurity standard from the production schedule of the API itself. In scenarios where API production is halted or scaled down, the ability to independently synthesize the impurity ensures that quality control testing can continue unabated. The robustness of the reaction conditions—operating at atmospheric pressure and moderate temperatures—means that the process can be easily transferred between different manufacturing sites or contract research organizations without requiring specialized high-pressure or cryogenic equipment. This flexibility enhances the resilience of the supply network, ensuring that regulatory compliance is maintained regardless of upstream production fluctuations.
- Scalability and Environmental Compliance: From an environmental and scalability standpoint, the process is well-suited for industrial adaptation. The reaction steps do not generate hazardous waste streams that require complex treatment; the byproducts are primarily salts and alcohols which are manageable within standard wastewater treatment protocols. The scalability is evidenced by the use of simple unit operations like extraction, washing, and distillation, which are easily amplified from gram-scale laboratory synthesis to kilogram-scale production. This ease of scale-up supports the growing regulatory demand for more comprehensive impurity profiling, allowing manufacturers to produce sufficient quantities of the reference standard to support long-term stability studies and multi-batch validation campaigns without encountering bottlenecks.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and technical specifications disclosed in the patent documentation, providing a clear understanding of the process capabilities and limitations. Understanding these details is essential for technical teams evaluating the feasibility of adopting this route for their internal reference standard production or for sourcing partners who utilize this methodology.
Q: Why is synthesizing this specific chlorpheniramine impurity critical for API manufacturers?
A: This impurity possesses a structure extremely close to chlorpheniramine maleate, making separation during purification difficult. Having a synthesized, high-purity reference standard allows manufacturers to accurately quantify and control this specific contaminant, ensuring regulatory compliance and product safety.
Q: What are the key reaction conditions for the synthesis described in CN111100067A?
A: The process utilizes relatively mild and scalable conditions. The initial alkylation occurs at 35-85°C in toluene; the hydrolysis step uses refluxing acetone with hydrochloric acid; and the final condensation proceeds at 0°C to room temperature in THF. These conditions avoid extreme pressures or cryogenic temperatures, facilitating easier scale-up.
Q: How does this synthetic route improve supply chain reliability for reference standards?
A: Traditional methods often rely on isolating trace impurities from bulk API, which is inefficient and yields inconsistent quantities. This dedicated synthetic route uses commercially available starting materials (such as p-chlorobenzonitrile and 2-chloropyridine derivatives) to produce the impurity on demand, ensuring a consistent and reliable supply of the reference standard independent of API production batches.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chlorpheniramine Impurity Supplier
The development of precise impurity standards like the one described in CN111100067A underscores the increasing complexity of modern pharmaceutical quality control. At NINGBO INNO PHARMCHEM, we recognize that access to high-purity reference materials is not just a regulatory requirement but a strategic asset for ensuring drug safety. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive materials that meet the most stringent purity specifications. Our rigorous QC labs are equipped to handle the complex analytical challenges associated with chiral and structural impurities, guaranteeing that every batch of material we supply is fully characterized and fit for purpose.
We invite pharmaceutical manufacturers and quality assurance professionals to collaborate with us to optimize their impurity management strategies. Whether you require custom synthesis of specific chlorpheniramine impurities or a comprehensive Customized Cost-Saving Analysis for your reference standard supply chain, our technical team is ready to assist. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments tailored to your project's unique requirements, ensuring your path to market is smooth, compliant, and efficient.