Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinone Derivatives for Commercial Scale-up
The pharmaceutical and fine chemical industries are constantly seeking robust methodologies to construct nitrogen-containing heterocycles, particularly quinazolinone derivatives, due to their profound biological activities ranging from anticancer to anticonvulsant properties. Patent CN112125856A introduces a groundbreaking preparation method for 2-trifluoromethyl substituted quinazolinone derivatives that addresses critical safety and efficiency bottlenecks found in traditional synthetic routes. This innovation leverages a transition metal palladium-catalyzed carbonylation tandem reaction, utilizing a solid carbon monoxide surrogate to eliminate the risks associated with gaseous CO. The strategic introduction of the trifluoromethyl group significantly enhances the metabolic stability and lipophilicity of the parent molecule, making these intermediates highly valuable for drug discovery programs targeting complex diseases. As a reliable pharmaceutical intermediate supplier, understanding the nuances of such patented technologies allows us to offer superior process solutions that align with modern regulatory and safety standards.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-trifluoromethyl substituted quinazolinone derivatives has been plagued by significant operational hazards and chemical inefficiencies that hinder large-scale adoption. Conventional literature reports often rely on the direct use of carbon monoxide gas, which is colorless, odorless, and highly toxic, necessitating expensive high-pressure reactors and rigorous safety protocols that drive up capital expenditure. Alternative methods involving cyclization with ethyl trifluoroacetate or trifluoroacetic anhydride frequently suffer from harsh reaction conditions, requiring extreme temperatures or strong acids that limit functional group tolerance. Furthermore, many existing pathways utilize expensive or pre-activated substrates that are not readily available in bulk quantities, leading to supply chain vulnerabilities and inconsistent batch-to-batch quality. These traditional approaches often result in low yields and narrow substrate scopes, making them economically unviable for the commercial scale-up of complex pharmaceutical intermediates required by global R&D teams.
The Novel Approach
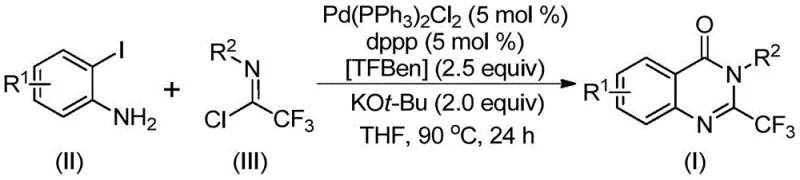
In stark contrast, the methodology disclosed in CN112125856A represents a paradigm shift by employing 1,3,5-tricarboxylate phenol ester (TFBen) as a safe, solid carbon monoxide substitute. This innovative approach allows the reaction to proceed under atmospheric pressure conditions, drastically simplifying the reactor requirements and enhancing operator safety without compromising reaction efficiency. The process utilizes cheap and easily obtainable starting materials, specifically trifluoroethylimidoyl chloride and o-iodoaniline, which are compatible with a wide variety of substituents including halogens, alkyl groups, and nitro groups. By operating at a moderate temperature of 90°C in common organic solvents like tetrahydrofuran, the method ensures high conversion rates while maintaining excellent functional group compatibility. This novel route not only widens the practical applicability of quinazolinone synthesis but also provides a streamlined pathway for cost reduction in pharmaceutical intermediate manufacturing by removing the need for specialized gas handling infrastructure.
Mechanistic Insights into Pd-Catalyzed Carbonylation Tandem Reaction
The mechanistic pathway of this transformation involves a sophisticated sequence of organometallic steps initiated by the base-promoted coupling of the starting materials. Initially, potassium tert-butoxide facilitates an intermolecular carbon-nitrogen bond coupling to generate a trifluoroacetamidine derivative in situ. Subsequently, the palladium catalyst inserts into the carbon-iodine bond of the o-iodoaniline moiety, forming a reactive divalent palladium intermediate that serves as the cornerstone of the catalytic cycle. Under the applied heating conditions, the solid TFBen decomposes to release carbon monoxide directly into the reaction medium, which then inserts into the carbon-palladium bond to form a crucial acyl-palladium intermediate. This precise control over CO release prevents the accumulation of toxic gas while ensuring a steady concentration for the carbonylation step. Finally, base-assisted promotion of palladium-nitrogen bond formation leads to a seven-membered ring palladium intermediate, which undergoes reductive elimination to yield the final 2-trifluoromethyl substituted quinazolinone derivative with high structural fidelity.
Controlling the impurity profile in such complex heterocyclic synthesis is paramount for meeting the stringent purity specifications required by regulatory bodies. The use of a solid CO surrogate inherently reduces the risk of side reactions often associated with uncontrolled gas flow rates in traditional carbonylation processes. The specific choice of ligands, such as 1,3-bis(diphenylphosphine)propane (dppp), stabilizes the palladium center and directs the regioselectivity of the insertion steps, minimizing the formation of regioisomers or homocoupling byproducts. Furthermore, the mild reaction conditions prevent the decomposition of sensitive functional groups on the aromatic rings, ensuring that the final impurity spectrum is clean and manageable. This level of mechanistic control is essential for R&D directors who require high-purity intermediates for downstream biological testing and clinical trial material production without extensive purification burdens.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
To implement this synthesis effectively, one must adhere to the precise stoichiometric ratios and environmental controls outlined in the patent data to maximize yield and reproducibility. The process begins with the careful weighing of the palladium catalyst, ligand, base, and the solid CO source, ensuring an inert atmosphere is maintained to protect the catalyst from oxidation. Detailed standardized synthesis steps see the guide below, which outlines the specific addition order and workup procedures necessary to isolate the product in high purity. Following the reaction period of 16 to 30 hours, the mixture requires filtration to remove inorganic salts, followed by silica gel treatment and column chromatography to achieve the desired pharmaceutical grade quality. Adhering to these parameters ensures that the commercial scale-up of complex pharmaceutical intermediates proceeds smoothly with minimal technical deviations.
- Combine palladium catalyst, dppp ligand, potassium tert-butoxide, TFBen solid CO source, trifluoroethylimidoyl chloride, and o-iodoaniline in an organic solvent such as THF.
- Heat the reaction mixture to 90°C and maintain stirring for a duration between 16 to 30 hours to ensure complete conversion.
- Upon completion, filter the mixture, mix with silica gel, and purify the crude product via column chromatography to isolate the target derivative.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this patented methodology offers substantial strategic advantages by fundamentally altering the cost structure and risk profile of producing quinazolinone derivatives. The elimination of toxic carbon monoxide gas removes the need for specialized storage facilities, gas cylinder logistics, and high-pressure safety valves, resulting in significant overhead cost savings for manufacturing sites. Additionally, the reliance on commercially available and stable solid reagents enhances supply chain reliability, as these materials are less susceptible to transportation restrictions compared to hazardous gases. This stability ensures consistent production schedules and reduces the lead time for high-purity intermediates, allowing pharmaceutical companies to accelerate their drug development timelines without supply bottlenecks. The operational simplicity also translates to reduced training requirements for plant operators and lower maintenance costs for reactor systems.
- Cost Reduction in Manufacturing: The substitution of gaseous carbon monoxide with a solid surrogate eliminates the capital investment required for high-pressure autoclaves and gas detection systems, leading to drastic simplification of the production infrastructure. By avoiding expensive pre-activated substrates and utilizing cheap starting materials like o-iodoaniline, the raw material cost per kilogram is significantly optimized. The high reaction efficiency and broad substrate scope mean that fewer batches are rejected due to poor conversion, further driving down the effective cost of goods sold. These factors combine to create a highly competitive pricing structure for clients seeking cost-effective routes for their API precursors.
- Enhanced Supply Chain Reliability: Sourcing solid chemical reagents is inherently more stable than managing the supply of compressed toxic gases, which are often subject to strict regulatory transport limitations and regional availability issues. The use of common organic solvents like tetrahydrofuran and readily available palladium catalysts ensures that the supply chain remains resilient against market fluctuations. This reliability is critical for supply chain heads who must guarantee continuous material flow for long-term commercial production campaigns. The robustness of the method against varying substituent groups also means that a single production line can be adapted for multiple derivatives, maximizing asset utilization.
- Scalability and Environmental Compliance: The absence of toxic gas emissions aligns perfectly with increasingly stringent environmental regulations, reducing the burden on waste treatment facilities and lowering compliance risks. The reaction operates at moderate temperatures and atmospheric pressure, making it inherently safer and easier to scale from laboratory grams to multi-ton commercial quantities without exponential increases in safety hazards. The post-treatment process involving standard filtration and chromatography is well-understood in the industry, facilitating easy technology transfer to contract manufacturing organizations. This scalability ensures that the method can support the growing demand for fluorinated heterocycles in the global pharmaceutical market.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology in an industrial setting. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation to ensure accuracy. Understanding these details helps stakeholders make informed decisions about adopting this route for their specific project needs. For further technical clarification, our team is available to discuss specific process parameters.
Q: What is the primary safety advantage of this synthesis method?
A: The method utilizes 1,3,5-tricarboxylate phenol ester (TFBen) as a solid carbon monoxide substitute, completely avoiding the use of toxic and hazardous carbon monoxide gas.
Q: What are the optimal reaction conditions described in the patent?
A: The reaction is optimally conducted at 90°C in tetrahydrofuran (THF) solvent for 16 to 30 hours using a palladium catalyst system with potassium tert-butoxide as the base.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the use of commercially available starting materials and the avoidance of high-pressure gas equipment make this protocol highly scalable and operationally simple for industrial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative patents like CN112125856A are translated into tangible supply solutions. Our facility is equipped with stringent purity specifications and rigorous QC labs capable of verifying the structural integrity and impurity profiles of complex fluorinated intermediates. We understand the critical nature of supply continuity for pharmaceutical clients and have optimized our processes to deliver high-purity 2-trifluoromethyl quinazolinone derivatives consistently. Our technical team is adept at troubleshooting scale-up challenges, ensuring that the transition from bench-scale discovery to commercial manufacturing is seamless and compliant with international standards.
We invite you to contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. By partnering with us, you gain access to specific COA data and route feasibility assessments that demonstrate the viability of this advanced synthesis method for your pipeline. Let us help you secure a stable, cost-effective, and safe supply of these critical building blocks for your next generation of therapeutic agents.