Advanced Synthesis of Nucleoside Intermediates for Scalable Pharmaceutical Manufacturing
Advanced Synthesis of Nucleoside Intermediates for Scalable Pharmaceutical Manufacturing
The pharmaceutical industry continuously seeks robust synthetic pathways that balance high purity with economic viability, particularly for complex nucleoside analogs used in antiviral and oncology therapies. Patent CN101805380B introduces a transformative approach for preparing (3S, 5S)-2,3-dihydroxy-5-hydroxymethyltetrahydrofuran triacetate, a pivotal intermediate in the synthesis of nucleoside drug parents. This innovation addresses critical bottlenecks in traditional manufacturing by replacing labor-intensive purification steps with efficient distillation protocols. By leveraging specific acid-base catalytic systems, the process achieves high yields while maintaining strict stereochemical control essential for biological activity. For global procurement teams, this represents a shift towards more sustainable and cost-effective supply chains for high-purity pharmaceutical intermediates. The methodology outlined in this patent provides a reliable foundation for scaling production from laboratory grams to multi-ton commercial quantities without compromising quality standards.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of this specific tetrahydrofuran derivative has relied heavily on silica gel column chromatography for product isolation, a technique that poses significant challenges for industrial scale-up. As documented in prior art such as Tetrahedron Vol.50, these conventional methods require vast quantities of organic solvents to elute the product, leading to excessive operational expenditures and environmental waste burdens. The complexity of column separation also introduces variability in batch-to-batch consistency, often resulting in lower overall recovery rates due to product retention on the stationary phase. Furthermore, the time-consuming nature of chromatographic purification extends the total production cycle, creating bottlenecks that hinder the ability to meet urgent market demands for antiviral ingredients. These factors collectively inflate the cost of goods sold, making the final API less competitive in price-sensitive global markets. Consequently, manufacturers have long sought an alternative that bypasses these inefficient downstream processing steps.
The Novel Approach
The patented methodology offers a streamlined solution by utilizing a direct acetylation strategy that simplifies the workup procedure to filtration and vacuum distillation. Instead of relying on chromatographic separation, the process exploits the differential solubility of byproducts, allowing impurities to precipitate out upon cooling while the target compound remains in the solution for distillation. This shift not only drastically reduces solvent consumption but also enables the recovery of valuable co-products, such as Compound C, which possesses its own industrial utility. The reaction conditions are remarkably mild yet effective, operating within a temperature range of 80°C to 160°C under inert gas protection to prevent degradation. By integrating protonic acids or Lewis bases as catalysts, the system ensures rapid conversion of the starting material, Compound A, into the desired triacetate structure. This approach aligns perfectly with green chemistry principles, offering a sustainable route that enhances atom economy and reduces the environmental footprint of nucleoside intermediate manufacturing.
Mechanistic Insights into Acid-Base Catalyzed Acetylation
The core of this synthetic breakthrough lies in the precise manipulation of reaction kinetics through the dual use of protonic acids and Lewis bases. The mechanism involves the activation of the hydroxyl groups on the tetrahydrofuran ring by the acetylating reagent, typically acetic anhydride or acetyl chloride, facilitated by catalysts like p-toluenesulfonic acid or DMAP. This catalytic environment promotes the formation of acetate esters at the 2, 3, and 5 positions with high regioselectivity. Crucially, the patent highlights that the stereochemical outcome at the 2-position of the furan ring is temperature-dependent, allowing chemists to tune the ratio of axial versus equatorial isomers. For instance, maintaining the reaction temperature above 145°C favors one configurational ratio, while lower temperatures shift the equilibrium, providing a valuable tool for optimizing the impurity profile. This level of control is vital for R&D directors who must ensure that the intermediate meets stringent specifications before coupling with nucleobases.
Understanding the upstream context is equally important for assessing the overall feasibility of the supply chain. The precursor, Compound A, is derived from adenosine through a series of protection and modification steps that establish the necessary deoxy-structure. 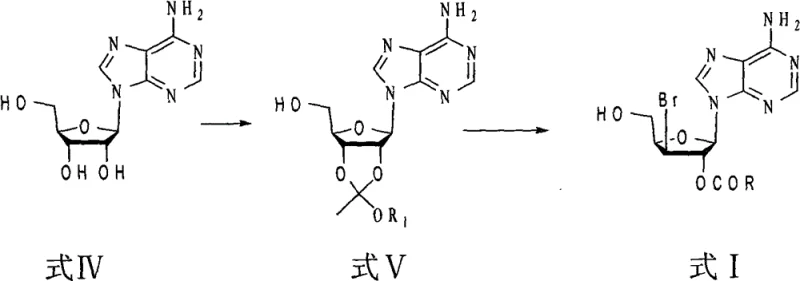 As illustrated in the reaction scheme, the transformation from natural adenosine involves orthoester formation followed by halogenation, setting the stage for the final acetylation step described in the main patent. This comprehensive view confirms that the entire synthetic lineage relies on commercially available starting materials and standard organic transformations. The robustness of these upstream steps ensures a consistent supply of high-quality Compound A, which is the critical feedstock for the patented acetylation process. By securing a reliable source for these precursors, manufacturers can mitigate supply chain risks and ensure continuous production of the final triacetate intermediate.
As illustrated in the reaction scheme, the transformation from natural adenosine involves orthoester formation followed by halogenation, setting the stage for the final acetylation step described in the main patent. This comprehensive view confirms that the entire synthetic lineage relies on commercially available starting materials and standard organic transformations. The robustness of these upstream steps ensures a consistent supply of high-quality Compound A, which is the critical feedstock for the patented acetylation process. By securing a reliable source for these precursors, manufacturers can mitigate supply chain risks and ensure continuous production of the final triacetate intermediate.
How to Synthesize (3S, 5S)-2,3-dihydroxy-5-hydroxymethyltetrahydrofuran triacetate Efficiently
Implementing this synthesis requires careful attention to reaction parameters to maximize yield and purity while minimizing side reactions. The process begins by dissolving Compound A in a polar organic solvent, such as acetic anhydride which serves a dual role as both reactant and medium. Catalysts are added in specific molar ratios, typically ranging from 1:0.05 to 1:0.2 for Lewis bases, to initiate the acetylation under nitrogen or argon atmosphere. Detailed standardized operating procedures for temperature ramping, reaction monitoring via HPLC, and the specific distillation cuts required to isolate the product are essential for reproducibility.
- React Compound A with an acetylating reagent like acetic anhydride in a polar organic solvent under inert gas protection.
- Utilize a protonic acid or Lewis base catalyst at elevated temperatures (80-160°C) to drive the reaction to completion.
- Isolate the target product via cooling, filtration of byproducts, and vacuum distillation to achieve high purity without column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented process translates into tangible strategic advantages beyond mere technical feasibility. The elimination of silica gel chromatography removes a major cost driver associated with stationary phase materials and the disposal of large volumes of hazardous solvent waste. This simplification of the downstream process significantly shortens the manufacturing lead time, allowing for faster response to market fluctuations and reduced inventory holding costs. Additionally, the ability to recover and utilize byproduct Compound C enhances the overall atom economy of the plant, turning what was once waste into a value-added stream. These efficiencies collectively contribute to a more resilient supply chain capable of sustaining long-term production contracts.
- Cost Reduction in Manufacturing: The removal of column chromatography drastically lowers the operational expenditure by reducing solvent usage and labor hours associated with complex purification. Without the need for expensive silica gel and the extensive solvent recycling infrastructure it requires, the variable cost per kilogram of the intermediate is significantly optimized. This cost efficiency allows pharmaceutical companies to maintain healthier margins or pass savings on to healthcare providers, enhancing the competitiveness of the final drug product in the global marketplace.
- Enhanced Supply Chain Reliability: By relying on simple unit operations like filtration and distillation, the process becomes less susceptible to the bottlenecks often caused by specialized purification equipment failures. The use of commercially available reagents and standard reactors means that production can be easily transferred between different manufacturing sites without extensive requalification. This flexibility ensures a continuous supply of high-purity pharmaceutical intermediates, mitigating the risk of stockouts that could delay critical drug development programs or commercial launches.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, moving seamlessly from laboratory flasks to industrial reactors without the geometric limitations of chromatography columns. The reduced solvent load simplifies waste treatment protocols, making it easier for facilities to comply with increasingly stringent environmental regulations regarding volatile organic compound emissions. This alignment with green chemistry standards not only future-proofs the manufacturing asset but also appeals to stakeholders prioritizing sustainability in their sourcing decisions.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These insights are derived directly from the experimental data and claims presented in the patent documentation to ensure accuracy.
Q: How does this new method improve upon traditional silica gel separation?
A: The patented method eliminates the need for costly and solvent-intensive silica gel column chromatography, replacing it with simple filtration and vacuum distillation which drastically reduces operational costs and waste.
Q: Can the stereochemistry of the furan ring be controlled during this process?
A: Yes, the ratio of axial to equatorial substituents at the 2-position of the furan ring can be modulated by adjusting the reaction temperature, allowing for optimization of the desired isomer profile.
Q: What are the primary applications of this tetrahydrofuran derivative?
A: This compound serves as a critical sugar-derived intermediate for synthesizing nucleoside drug parents, specifically used in the development of antibiotics, anti-AIDS medications, and cancer diagnostic agents.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable (3S, 5S)-2,3-dihydroxy-5-hydroxymethyltetrahydrofuran triacetate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the successful development of next-generation antiviral and oncology therapies. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met with precision and reliability. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of (3S, 5S)-2,3-dihydroxy-5-hydroxymethyltetrahydrofuran triacetate meets the highest industry standards. Our commitment to technical excellence allows us to navigate complex synthetic challenges effectively, delivering materials that facilitate smooth downstream processing for our partners.
We invite you to engage with our technical procurement team to discuss how our manufacturing capabilities can support your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into how our optimized processes can reduce your overall cost of goods. We encourage potential partners to contact us directly to obtain specific COA data and route feasibility assessments tailored to your unique development timeline and quality expectations.