Advancing Pharmaceutical Intermediate Production via Green Electrochemical Cyclization Technology
Advancing Pharmaceutical Intermediate Production via Green Electrochemical Cyclization Technology
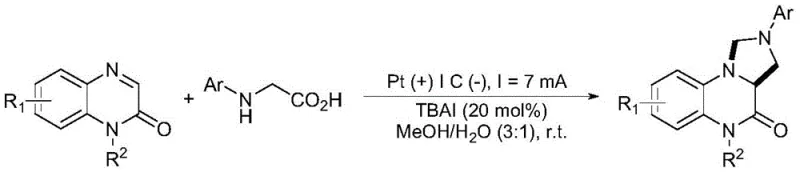
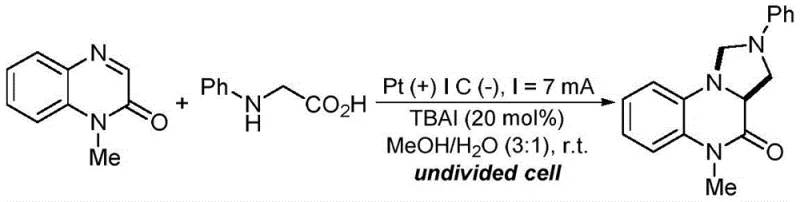
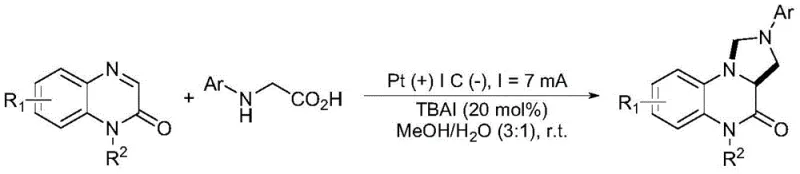
The landscape of pharmaceutical intermediate manufacturing is undergoing a significant transformation driven by the urgent need for greener, more sustainable, and cost-effective synthetic methodologies. A pivotal advancement in this domain is documented in patent CN115637454A, which discloses a novel electrochemical method for synthesizing tetrahydroimidazo[1,5-a]quinoxalin-4(5H)-one derivatives. These heterocyclic scaffolds are critical pharmacophores found in numerous bioactive compounds with anticancer, antitumor, and antibacterial properties. The patented technology leverages direct current electrolysis to facilitate an oxidative decarboxylation coupling-cyclization reaction between quinoxalin-2(1H)-ones and N-arylglycines. By utilizing electrons as a traceless oxidant and inexpensive iodide salts as mediators, this approach eliminates the reliance on stoichiometric chemical oxidants and precious metal photocatalysts, marking a substantial leap forward in process chemistry efficiency and environmental stewardship for the fine chemical industry.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of tetrahydroimidazo[1,5-a]quinoxalin-4(5H)-one frameworks has relied heavily on transition-metal catalysis or photocatalytic systems that present inherent logistical and economic challenges. Previous literature, such as studies utilizing CsPbBr3 or graphitic carbon nitride (g-C3N4) under visible light irradiation, while innovative, suffers from significant drawbacks regarding industrial scalability. These photocatalytic protocols often necessitate the use of expensive, air-sensitive, or toxic catalysts that are difficult to recover and recycle, thereby inflating the cost of goods sold (COGS). Furthermore, the requirement for specialized light sources and the limited penetration of light in large-scale reactors create bottlenecks in throughput. The functional group tolerance in these older methods is also frequently compromised, leading to lower yields when complex substrates bearing sensitive moieties are employed, which ultimately complicates downstream purification and reduces overall process atom economy.
The Novel Approach
In stark contrast, the electrochemical strategy outlined in the patent offers a robust alternative that directly addresses these pain points by replacing chemical reagents with electrical energy. This method operates in an undivided cell using a simple combination of a platinum anode and a graphite cathode, driven by a constant direct current. The core innovation lies in the synergistic catalysis provided by organic quaternary ammonium salts or inorganic iodides, such as tetrabutylammonium iodide (TBAI), which mediate the electron transfer processes efficiently. This setup allows the reaction to proceed at room temperature in a mixed solvent system of methanol and water, drastically simplifying the operational requirements. By avoiding the use of external chemical oxidants, the process generates fewer by-products and waste streams, aligning perfectly with the principles of green chemistry while maintaining high reaction selectivity and yield, making it an ideal candidate for reliable pharmaceutical intermediate supplier operations.
Mechanistic Insights into Electrochemical Oxidative Decarboxylation Cyclization
The mechanistic pathway of this transformation is a sophisticated interplay of anodic oxidation and cathodic reduction events mediated by iodide species. In the electrolyte solution, the N-arylglycine substrate interacts with water molecules to form a hydrogen-bonded complex, which is subsequently reduced at the cathode surface to generate a radical anion intermediate while releasing hydrogen gas. Simultaneously, at the anode, iodide ions are oxidized to molecular iodine or iodine radicals. These active iodine species then oxidize the glycine-derived intermediate to form an unstable hypoiodite species, which rapidly undergoes homolytic cleavage to produce an oxygen-centered radical. This radical swiftly decarboxylates to yield a crucial carbon-centered aminomethyl radical. This highly reactive species selectively attacks the C3 position of the quinoxalin-2(1H)-one ring, initiating a cascade of radical coupling and intramolecular cyclization events that ultimately lead to the formation of the fused imidazo-quinoxaline core with the release of an aniline by-product, demonstrating a highly controlled radical manifold.
Understanding the impurity profile is critical for R&D directors focused on regulatory compliance, and this electrochemical mechanism offers distinct advantages in this regard. The use of electricity as the primary oxidant ensures that the oxidation potential can be finely tuned by adjusting the current density, thereby minimizing over-oxidation side reactions that are common with harsh chemical oxidants like peroxides or hypervalent iodine reagents. The selective generation of the aminomethyl radical ensures that the coupling occurs specifically at the desired C3 position of the quinoxaline ring, reducing the formation of regioisomers. Furthermore, the mild aqueous-organic solvent system helps to solubilize polar intermediates and salts, preventing the precipitation of tarry by-products that often plague non-polar organic syntheses. This results in a cleaner crude reaction mixture, which significantly reduces the burden on purification teams and enhances the overall purity of the final high-purity pharmaceutical intermediate.
How to Synthesize Tetrahydroimidazo[1,5-a]quinoxalin-4(5H)-one Efficiently
Implementing this electrochemical protocol in a laboratory or pilot plant setting requires careful attention to electrode configuration and electrolyte composition to maximize efficiency. The standard procedure involves dissolving the quinoxalin-2(1H)-one and N-arylglycine substrates in a methanol-water mixture (typically 3:1 ratio) along with 20 mol% of TBAI as the supporting electrolyte and catalyst. The reaction is conducted in an undivided cell equipped with a platinum plate anode and a graphite plate cathode, applying a constant current of 7 mA at room temperature. Monitoring the reaction progress via thin-layer chromatography (TLC) is recommended, with typical reaction times ranging from 3 to 8 hours depending on the specific substrate electronics. For detailed operational parameters and safety guidelines, please refer to the standardized synthesis steps provided below.
- Prepare the electrolyte solution by mixing quinoxalin-2(1H)-one, N-arylglycine, and tetrabutylammonium iodide (TBAI) in a methanol/water solvent system.
- Assemble an undivided electrochemical cell equipped with a platinum anode and a graphite cathode, ensuring proper electrode immersion.
- Apply a constant direct current of 7 mA at room temperature for approximately 5 hours, monitoring progress via TLC until completion.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this electrochemical methodology represents a strategic opportunity to optimize cost structures and mitigate supply risks associated with complex organic synthesis. The elimination of expensive photocatalysts and stoichiometric oxidants directly translates to a reduction in raw material expenditure, as the primary reagents are commodity chemicals like amino acid derivatives and simple quinoxalinones. Moreover, the operational simplicity of running reactions at ambient temperature without the need for cryogenic cooling or high-pressure equipment lowers the energy consumption and capital investment required for reactor infrastructure. This streamlined process flow enhances the reliability of the supply chain by reducing the number of unit operations and minimizing the potential for batch-to-batch variability caused by sensitive reagent handling.
- Cost Reduction in Manufacturing: The economic benefits of this technology are driven primarily by the substitution of high-cost catalytic systems with inexpensive iodide salts and electricity. By removing the need for precious metal photocatalysts like cesium lead bromide, manufacturers can avoid the volatility associated with rare earth metal pricing and the complex logistics of catalyst recovery. Additionally, the high atom efficiency of the decarboxylative coupling means that less starting material is wasted, further driving down the effective cost per kilogram of the active pharmaceutical ingredient (API) intermediate. The simplified workup procedure, which often requires only concentration and column chromatography, reduces solvent usage and labor hours, contributing to substantial overall cost savings in the manufacturing budget.
- Enhanced Supply Chain Reliability: Sourcing stability is a critical concern for global supply chains, and this method improves resilience by relying on widely available bulk chemicals rather than specialized reagents with long lead times. N-arylglycines and quinoxalinones are commercially accessible building blocks with established supply networks, reducing the risk of production stoppages due to material shortages. The robustness of the electrochemical conditions, which tolerate a wide range of functional groups including halogens and electron-withdrawing groups, ensures that diverse analogues can be produced using the same standard operating procedures. This flexibility allows for rapid scale-up and adaptation to changing market demands without the need for extensive process re-validation or re-tooling of production lines.
- Scalability and Environmental Compliance: Scaling electrochemical reactions is increasingly feasible with modern flow chemistry technologies, allowing for the seamless transition from gram-scale discovery to multi-ton commercial production. The absence of hazardous chemical oxidants significantly reduces the generation of toxic waste streams, simplifying wastewater treatment and ensuring compliance with stringent environmental regulations. The use of water as a co-solvent not only improves the sustainability profile of the process but also enhances safety by reducing the flammability risks associated with purely organic solvent systems. These factors collectively make the process highly attractive for long-term commercial scale-up of complex pharmaceutical intermediates in a regulated environment.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating the feasibility of this technology for their specific projects, we have compiled answers to common inquiries based on the patent data. These responses address key concerns regarding reaction scope, safety, and scalability, providing a clear picture of the operational benefits. Understanding these technical nuances is essential for making informed decisions about integrating electrochemical synthesis into your existing manufacturing portfolio.
Q: What are the advantages of this electrochemical method over traditional photocatalysis?
A: Unlike photocatalytic methods requiring expensive and unstable catalysts like CsPbBr3 or g-C3N4, this electrochemical approach uses electrons as a traceless oxidant and inexpensive iodide salts, significantly reducing raw material costs and simplifying product purification.
Q: Does this process require harsh reaction conditions or external oxidants?
A: No, the reaction proceeds under mild conditions at room temperature without the need for hazardous chemical oxidants. The use of electricity as the driving force enhances safety and environmental compliance for large-scale manufacturing.
Q: What is the substrate compatibility for this synthesis route?
A: The method demonstrates excellent functional group tolerance, successfully accommodating various substituents such as halogens, alkyl groups, alkoxy groups, and trifluoromethyl groups on both the quinoxaline and glycine components with high yields.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tetrahydroimidazo[1,5-a]quinoxalin-4(5H)-one Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of electrochemical synthesis in modern drug development and are committed to bringing these innovations to our global partners. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory benchtop to industrial reactor is seamless and efficient. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of tetrahydroimidazo[1,5-a]quinoxalin-4(5H)-one derivative meets the highest quality standards required for clinical and commercial applications. Our state-of-the-art facilities are equipped to handle complex electrochemical processes safely and effectively, positioning us as a trusted partner for your most challenging synthetic needs.
We invite you to collaborate with us to leverage this cutting-edge technology for your next project. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our advanced electrochemical capabilities can accelerate your timeline and reduce your overall manufacturing costs. Let us help you secure a stable, high-quality supply of critical pharmaceutical intermediates for your pipeline.