Advanced Manufacturing of 4-Chloro-N-methylpicolinamide for Scalable Sorafenib Production
Introduction to Next-Generation Sorafenib Intermediate Manufacturing
The pharmaceutical landscape for multi-kinase inhibitors continues to evolve, driven by the demand for cost-effective and high-purity intermediates. Patent CN102675197A, published in September 2012, introduces a transformative methodology for synthesizing 4-chloro-N-methylpicolinamide, a critical building block for the anti-tumor agent Sorafenib. This patent addresses long-standing challenges in heterocyclic chemistry, specifically the balance between reactivity and selectivity during amidation. By shifting from traditional organic solvent-based systems to an aqueous-mediated protocol, the disclosed technology offers a robust pathway that significantly enhances both yield and environmental profile. For R&D directors and supply chain leaders, this represents a pivotal opportunity to optimize the production of complex pyridine derivatives.
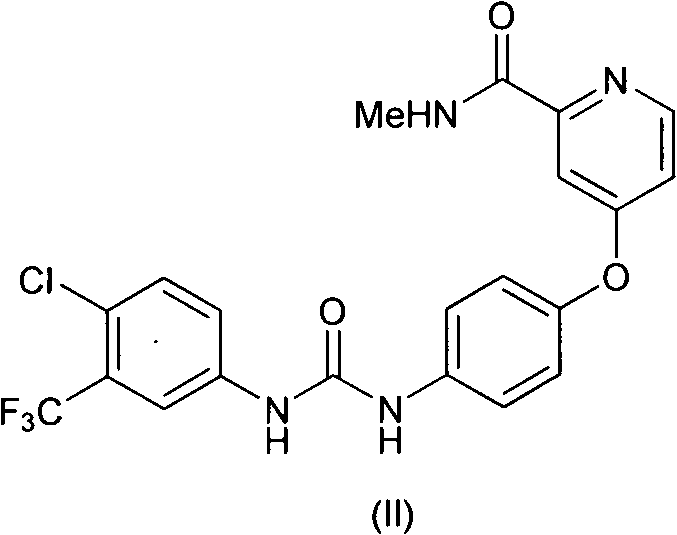
The core innovation lies in the strategic manipulation of the pyridine ring's reactivity. Traditional methods often struggle with the competing nucleophilic attacks that degrade product quality. The inventors have successfully decoupled these reaction pathways, ensuring that the methylamine moiety attaches exclusively to the carbonyl carbon rather than displacing the crucial chloro-substituent on the aromatic ring. This level of control is essential for maintaining the stringent purity specifications required for oncology APIs. As we delve deeper into the technical specifics, it becomes clear that this process is not merely an incremental improvement but a fundamental rethinking of the synthetic strategy for this class of compounds.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies for synthesizing 4-chloro-N-methylpicolinamide typically rely on two distinct but flawed approaches. The first method utilizes methylamine in organic solutions to react with ester intermediates. However, organic amines are potent nucleophiles that do not discriminate effectively between the ester carbonyl and the electron-deficient chloro-pyridine ring. This lack of selectivity leads to the formation of significant impurities, specifically 4-methylamino-pyridine-2-formyl-(N-methyl) amine, via nucleophilic aromatic substitution. Removing these structurally similar by-products is notoriously difficult and costly, often requiring extensive chromatography or recrystallization cycles that destroy overall yield. Furthermore, the heavy reliance on organic solvents inflates production costs and creates substantial waste disposal burdens, conflicting with modern green chemistry mandates.
The second conventional approach attempts to bypass the ester step by reacting the acid chloride directly with aqueous methylamine. While this avoids organic solvents, it introduces a different catastrophic failure mode: hydrolysis. Acid chlorides are extremely sensitive to water, and in an aqueous environment, the rate of hydrolysis to the corresponding carboxylic acid often outcompetes the desired amidation. This results in low conversion rates and the accumulation of acidic by-products that complicate downstream purification. Additionally, the resulting amide product often has a low melting point, making isolation from the reaction mixture challenging. These combined factors—impurity generation in organic media and hydrolysis in aqueous media—have historically created a bottleneck in the reliable supply of high-quality Sorafenib intermediates.
The Novel Approach
The methodology disclosed in CN102675197A elegantly resolves these contradictions by introducing a stabilized ester intermediate coupled with a controlled aqueous amidation step. Instead of reacting the highly reactive acid chloride directly with water, the process first converts it into a 4-chloropyridine-2-methyl formate hydrochloride salt. This esterification step effectively 'masks' the high reactivity of the acid chloride, rendering it stable enough to withstand the subsequent aqueous environment without undergoing rapid hydrolysis. Crucially, the use of aqueous methylamine (30-40% concentration) provides a unique reactivity profile. The presence of water solvates the amine, slightly reducing its nucleophilicity just enough to prevent attack on the aromatic ring chlorine, while retaining sufficient strength to cleave the ester bond and form the amide. This delicate balance allows the reaction to proceed with exceptional selectivity.
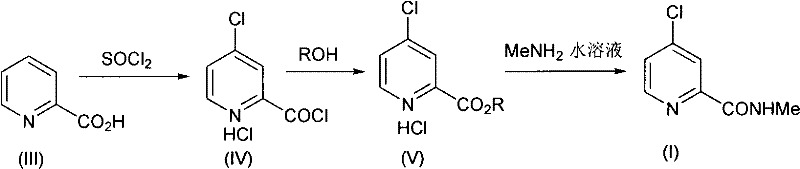
Furthermore, the novel approach eliminates the need for organic solvents in the final amidation step. By performing the reaction in water, the process drastically reduces the E-factor (environmental factor) of the synthesis. The product, being less soluble in water than the reagents, can often be isolated through simple extraction or precipitation, avoiding the energy-intensive distillation of organic solvents. Experimental data from the patent indicates that this route achieves yields as high as 98% with purities exceeding 98%, a significant leap over the variable and often lower performance of legacy methods. This robustness makes the process ideally suited for industrial scale-up, offering a reliable [pharmaceutical intermediates] supplier pathway that aligns with both economic and regulatory goals.
Mechanistic Insights into Selective Amidation and Impurity Control
To fully appreciate the technical depth of this invention, one must analyze the electronic and steric factors governing the reaction mechanism. The pyridine ring in 4-chloropyridine derivatives is electron-deficient due to the electronegative nitrogen atom, which activates the ring towards nucleophilic aromatic substitution (SnAr), particularly at the 4-position where the chlorine resides. In organic solvents, free methylamine acts as a 'hard' nucleophile with high energy, easily overcoming the activation barrier to displace the chloride ion. However, in the aqueous system proposed by the patent, the hydrogen bonding network of water molecules forms a solvation shell around the methylamine. This solvation effect lowers the ground state energy of the nucleophile, effectively dampening its aggression towards the aromatic ring. Consequently, the activation energy for SnAr becomes prohibitive under the mild reaction temperatures (0-40°C) specified.
Simultaneously, the ester intermediate plays a critical role in kinetic control. Unlike the acid chloride, which reacts instantaneously and indiscriminately with water, the methyl ester possesses a higher activation energy for hydrolysis. This kinetic stability provides a time window wherein the aminolysis reaction—the attack of the amine on the carbonyl carbon—can occur preferentially. The mechanism likely proceeds through a tetrahedral intermediate where the methylamine attacks the carbonyl, followed by the elimination of methanol. Because methanol is a better leaving group in this specific context than the chloride on the ring, and because the aqueous environment suppresses the competing SnAr pathway, the reaction funnel is directed almost exclusively toward the desired amide product. This mechanistic understanding underscores why the specific combination of an ester precursor and aqueous amine is the key to unlocking high purity.
From an impurity control perspective, this mechanism inherently minimizes the formation of the dreaded 4-methylamino by-product. In traditional organic syntheses, this impurity is often generated in quantities that require specialized scavengers or complex workups to remove. By suppressing its formation at the source, the new process simplifies the purification train. The patent examples demonstrate that simple liquid-liquid extraction with ethyl acetate is sufficient to isolate the product with 98% purity. This suggests that the impurity profile is clean, lacking the complex array of side products seen in acid chloride hydrolysis or organic aminolysis. For quality control teams, this translates to more consistent batch-to-batch reproducibility and reduced risk of failing specification limits for genotoxic impurities or related substances.
How to Synthesize 4-Chloro-N-methylpicolinamide Efficiently
The synthesis of this critical intermediate is streamlined into three operational steps that maximize throughput while minimizing handling hazards. The process begins with the activation of picolinic acid using thionyl chloride, followed by a telescoped esterification, and concludes with the innovative aqueous amidation. This sequence is designed to avoid the isolation of unstable intermediates, thereby enhancing safety and efficiency. The detailed standardized synthesis steps, including precise stoichiometric ratios and temperature ramps, are outlined in the guide below to ensure reproducible results in your pilot or production facilities.
- React pyridine-2-formic acid with thionyl chloride and DMF catalyst to form 4-chloropyridine-2-formyl chloride hydrochloride.
- Perform esterification on the crude acid chloride using methanol and toluene to generate 4-chloropyridine-2-methyl formate hydrochloride.
- React the ester intermediate with 30-40% aqueous methylamine solution at 0-40°C without organic solvents to yield the final amide.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented methodology offers compelling strategic advantages beyond mere technical superiority. The shift towards aqueous chemistry fundamentally alters the cost structure of manufacturing this intermediate. By eliminating the requirement for expensive, anhydrous organic solvents in the final coupling step, the process reduces raw material expenditure significantly. Moreover, the removal of organic solvents simplifies the recovery and recycling infrastructure needed on-site. There is no need for large-scale distillation columns to recover amines or solvents from the final reaction mass, which lowers both capital expenditure (CAPEX) for new plants and operational expenditure (OPEX) for existing ones. This efficiency drives substantial cost reduction in pharmaceutical intermediates manufacturing, allowing for more competitive pricing in the global market.
- Cost Reduction in Manufacturing: The economic benefits of this route are multifaceted. Firstly, the use of aqueous methylamine is inherently cheaper than handling specialized organic amine solutions, which often require stabilizers and rigorous moisture control. Secondly, the high yield (up to 98%) means that less starting material is wasted, directly improving the atom economy of the process. In a commercial setting, a yield increase from a typical 70-80% to nearly quantitative levels translates to massive savings in raw material costs per kilogram of API produced. Additionally, the simplified workup—often requiring only extraction and evaporation rather than column chromatography—reduces labor hours and consumable costs like silica gel or specialized resins. These factors combine to create a leaner, more profitable manufacturing model.
- Enhanced Supply Chain Reliability: Supply continuity is paramount for blockbuster drugs like Sorafenib. This process enhances reliability by utilizing commodity chemicals that are widely available globally. Thionyl chloride, methanol, and aqueous methylamine are bulk commodities with stable supply chains, unlike specialized catalysts or exotic reagents that might be sourced from single suppliers. The robustness of the reaction conditions (0-40°C) also means that the process is less sensitive to minor fluctuations in utility supplies, such as cooling water temperature variations. This resilience reduces the risk of batch failures due to operational upsets, ensuring a steady flow of [high-purity pharmaceutical intermediates] to downstream API manufacturers. The ability to run the reaction without strict anhydrous conditions further de-risks the supply chain by removing the need for complex drying protocols.
- Scalability and Environmental Compliance: As regulatory pressure mounts regarding solvent emissions and waste disposal, this aqueous-based process positions manufacturers favorably for the future. The drastic reduction in volatile organic compound (VOC) emissions during the final step simplifies compliance with environmental regulations. Waste streams are primarily aqueous and saline, which are easier and cheaper to treat than mixed organic waste. From a scalability standpoint, the exotherm of the amidation reaction is manageable in water due to the high heat capacity of the solvent, reducing the risk of thermal runaway during scale-up from kilograms to tons. This safety profile facilitates faster technology transfer and quicker ramp-up times, enabling suppliers to respond rapidly to surges in market demand without compromising safety or quality standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These insights are derived directly from the experimental data and claims within the patent documentation, providing a clear picture of what partners can expect when adopting this technology. Understanding these nuances is critical for making informed decisions about process validation and vendor qualification.
Q: Why is aqueous methylamine preferred over organic methylamine for this synthesis?
A: Using organic methylamine solutions can lead to nucleophilic aromatic substitution on the pyridine ring, creating difficult-to-remove impurities. Aqueous methylamine mitigates this side reaction while maintaining sufficient nucleophilicity for amidation.
Q: How does this process improve yield compared to traditional acid chloride routes?
A: Traditional routes reacting acid chlorides directly with aqueous amines suffer from rapid hydrolysis of the acid chloride group. Converting to an ester intermediate first stabilizes the molecule against hydrolysis, allowing for high-yield conversion to the amide.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the process eliminates the need for organic solvents in the final step and utilizes cheap, readily available reagents like thionyl chloride and aqueous methylamine, making it highly cost-effective and environmentally friendly for ton-scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 4-Chloro-N-methylpicolinamide Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory patent to commercial reality requires more than just chemical knowledge; it demands engineering excellence. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of CN102675197A are fully realized in practice. Our facilities are equipped with rigorous QC labs capable of detecting trace impurities at ppm levels, guaranteeing that every batch meets stringent purity specifications required for oncology applications. We understand the critical nature of Sorafenib intermediates in the global supply chain and are committed to delivering consistency and quality.
We invite you to collaborate with us to leverage this advanced synthetic route for your supply needs. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating exactly how this greener process impacts your bottom line. Please contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us partner with you to secure a sustainable and cost-efficient supply of this vital pharmaceutical building block.