Advanced Synthesis of Istradefylline Demethylated Impurities for Global Pharmaceutical Quality Control
Advanced Synthesis of Istradefylline Demethylated Impurities for Global Pharmaceutical Quality Control
The pharmaceutical industry faces increasing regulatory pressure to comprehensively characterize the impurity profiles of active pharmaceutical ingredients (APIs), particularly for novel therapies targeting neurological disorders. Patent CN113248505B introduces a groundbreaking preparation method for three specific demethylated impurities of Istradefylline, a potent adenosine A2a receptor antagonist used in the treatment of Parkinson's disease. This technology addresses a critical gap in the supply chain by providing a robust, scalable route to generate high-purity reference standards that are essential for rigorous quality control and safety evaluation. By utilizing a controlled demethylation strategy followed by selective alkylation, this process ensures that pharmaceutical manufacturers can accurately quantify trace degradation products, thereby safeguarding patient safety and meeting stringent global regulatory requirements for impurity identification.
As the global demand for Parkinson's disease treatments rises, the ability to reliably source certified reference materials becomes a bottleneck for many generic and innovator drug companies. The methodology outlined in this patent not only simplifies the synthesis of these complex heterocyclic compounds but also significantly enhances the purity profile compared to traditional extraction or non-specific synthesis routes. For procurement leaders and supply chain managers, understanding the technical nuances of this synthesis is vital, as it represents a shift towards more predictable and cost-effective sourcing of critical analytical standards. This report delves into the mechanistic details and commercial implications of adopting this advanced preparation technique for securing a stable supply of high-quality pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, obtaining specific positional isomers of demethylated xanthine derivatives like Istradefylline impurities has been fraught with challenges related to regioselectivity and purification complexity. Conventional synthetic routes often rely on harsh conditions that lead to indiscriminate demethylation, resulting in complex mixtures of mono-demethylated and di-demethylated species that are difficult to separate. Furthermore, existing methods frequently suffer from low yields and require extensive chromatographic purification, which drives up the cost of goods and limits the availability of these materials for routine quality control testing. The lack of standardized, high-purity reference substances has historically hindered the ability of pharmaceutical companies to fully validate their analytical methods, creating risks during regulatory audits and potentially delaying product launches due to insufficient impurity characterization data.
The Novel Approach
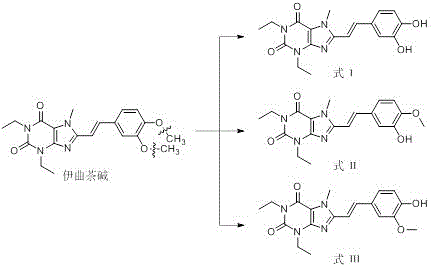
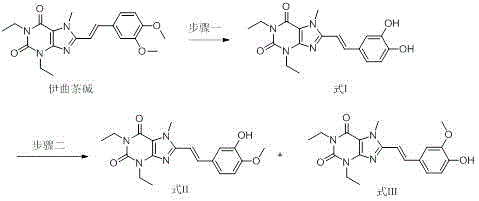
The innovative process disclosed in patent CN113248505B overcomes these historical hurdles by employing a stepwise, controlled chemical transformation that prioritizes both yield and selectivity. Instead of attempting to synthesize the impurities from scratch, the method cleverly utilizes Istradefylline itself as the starting material, subjecting it to specific hydrobromic acid hydrolysis to generate the di-hydroxy intermediate (Formula I) with high efficiency. This intermediate is then subjected to a controlled methylation reaction using methyl iodide and a mild acid-binding agent, which allows for the selective formation of the mono-methoxy isomers (Formula II and Formula III). This strategic approach minimizes side reactions and simplifies the downstream purification process, ultimately delivering reference standards with purity levels exceeding 98% through a streamlined workflow that is far more amenable to industrial scale-up than previous art.
Mechanistic Insights into Acid-Catalyzed Demethylation and Selective Alkylation
The core of this technological advancement lies in the precise manipulation of ether cleavage and nucleophilic substitution mechanisms within the purine scaffold. In the first stage, the reaction utilizes concentrated hydrobromic acid at elevated temperatures (around 120°C) to effect the cleavage of the methoxy groups on the phenyl ring. This acid-catalyzed hydrolysis proceeds via a protonation of the ether oxygen followed by nucleophilic attack by the bromide ion, effectively converting the 3,4-dimethoxyphenyl group into a 3,4-dihydroxyphenyl moiety without degrading the sensitive xanthine core. The reaction conditions are carefully optimized to ensure complete conversion while preventing the hydrolysis of the amide bonds within the purine ring, a critical balance that ensures the structural integrity of the resulting Formula I intermediate is maintained for subsequent transformations.
Following the isolation of the di-hydroxy intermediate, the second stage involves a regioselective alkylation to reintroduce a single methyl group, thereby generating the specific mono-demethylated isomers required for impurity profiling. By controlling the molar ratio of methyl iodide and utilizing potassium carbonate as a base in polar aprotic solvents like DMF or DCM, the reaction favors the formation of the mono-methylated products over the fully remethylated starting material. The subtle differences in the electronic environment of the two hydroxyl groups allow for the formation of both the 3-methoxy-4-hydroxy and 3-hydroxy-4-methoxy isomers. The final separation is achieved through preparative liquid chromatography, leveraging the slight polarity differences between the isomers to isolate Formula II and Formula III with exceptional purity, ensuring they are fit for purpose as analytical standards.
How to Synthesize Istradefylline Demethylated Impurities Efficiently
The synthesis of these critical reference standards requires strict adherence to the reaction parameters defined in the patent to ensure reproducibility and high purity. The process begins with the thermal reaction of Istradefylline with hydrobromic acid, followed by a crystallization step to isolate the di-hydroxy intermediate. Subsequent methylation and chromatographic separation yield the final isomers. For detailed operational parameters, stoichiometry, and purification gradients, please refer to the structured guide below which outlines the standard operating procedure derived from the patent examples.
- React Istradefylline with hydrobromic acid solution at elevated temperatures to effect demethylation, yielding the di-hydroxy compound (Formula I).
- Purify the crude Formula I product via recrystallization using N,N-dimethylformamide and water to achieve high purity.
- Subject Formula I to selective methylation using methyl iodide and an acid-binding agent in organic solvent, followed by preparative liquid chromatography to separate Formula II and Formula III.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route offers significant strategic advantages beyond mere technical feasibility. The ability to produce these impurities in-house or source them from a supplier utilizing this efficient method translates directly into reduced dependency on scarce external vendors who may charge premium prices for small quantities of reference standards. By simplifying the purification process and improving overall yields, the cost of producing these high-value analytical materials is substantially lowered, allowing pharmaceutical companies to allocate resources more effectively across their R&D portfolios. Furthermore, the robustness of the method ensures a consistent supply of materials, mitigating the risk of project delays caused by the unavailability of critical impurity standards during method validation and stability studies.
- Cost Reduction in Manufacturing: The streamlined nature of this synthesis eliminates the need for complex multi-step sequences often required to build the purine skeleton from scratch, thereby reducing raw material consumption and processing time. By using the API itself as the starting material, the process maximizes atom economy for the specific impurity targets, leading to significant operational cost savings in the production of reference standards. Additionally, the simplified purification protocol reduces the consumption of expensive chromatography columns and solvents, further driving down the total cost of ownership for quality control laboratories seeking to maintain comprehensive impurity libraries.
- Enhanced Supply Chain Reliability: Relying on a synthesis method that utilizes readily available reagents like hydrobromic acid and methyl iodide ensures that production is not vulnerable to the supply fluctuations of exotic catalysts or specialized precursors. This chemical robustness guarantees a steady flow of materials, enabling procurement teams to negotiate better terms and secure long-term contracts with confidence. The high yield and purity reported in the patent examples suggest that suppliers can maintain healthy inventory levels, drastically reducing lead times for urgent orders and ensuring that pharmaceutical development timelines are not compromised by material shortages.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing common organic solvents and standard reaction conditions that can be easily transferred from laboratory to pilot and commercial scales. The avoidance of heavy metal catalysts and the use of straightforward workup procedures align with modern green chemistry principles, reducing the environmental footprint associated with waste disposal. This compliance with environmental regulations simplifies the permitting process for manufacturing facilities and ensures that the supply chain remains resilient against tightening global environmental standards, making it a sustainable choice for long-term procurement strategies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these Istradefylline impurities. These answers are derived directly from the technical specifications and beneficial effects described in the patent documentation, providing clarity for stakeholders evaluating the implementation of this technology.
Q: What is the primary advantage of this preparation method for Istradefylline impurities?
A: The method described in patent CN113248505B offers a simple, high-yield process that produces impurities with purity exceeding 98%, solving the market shortage of qualified reference standards for quality control.
Q: How are the regioisomeric impurities (Formula II and III) separated?
A: After the selective methylation reaction, the mixture containing Formula II and Formula III is separated using preparative liquid phase chromatography with a specific gradient of acetonitrile and water, ensuring high isolation purity.
Q: Why are these specific demethylated impurities critical for drug development?
A: These impurities represent potential degradation products formed during synthesis or storage; having certified reference standards allows manufacturers to strictly monitor impurity profiles, ensuring the safety and efficacy of the final Parkinson's disease medication.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Istradefylline Impurities Supplier
At NINGBO INNO PHARMCHEM, we recognize that the integrity of your pharmaceutical products depends on the quality of your analytical standards. As a leading CDMO expert, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that even complex heterocyclic impurities like those described in CN113248505B are manufactured with precision. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of Istradefylline demethylated impurities we supply meets the highest international standards for identity and purity.
We invite you to collaborate with us to optimize your supply chain for Parkinson's disease therapeutics. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how our efficient synthesis routes can reduce your overall procurement costs. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us support your commitment to delivering safe and effective medicines to patients worldwide.