Advanced Pd-Catalyzed Decarboxylative Cyclization for Commercial Scale-Up of Complex Pharmaceutical Intermediates
Advanced Pd-Catalyzed Decarboxylative Cyclization for Commercial Scale-Up of Complex Pharmaceutical Intermediates
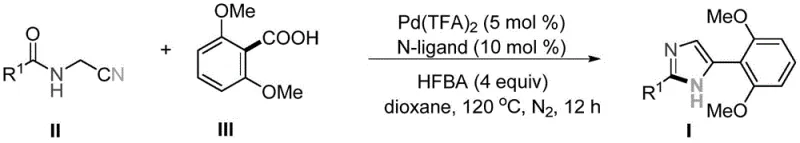
The development of efficient synthetic routes for nitrogen-containing heterocycles remains a critical priority in modern medicinal chemistry, particularly for scaffolds found in antiviral therapeutics. Patent CN113582929B introduces a groundbreaking methodology for the synthesis of 2,5-disubstituted imidazole compounds, a core structural motif essential for drugs like the chronic hepatitis C antiviral BMS-790052. This innovation addresses long-standing challenges in heterocyclic construction by leveraging a palladium-catalyzed decarboxylative tandem cyclization strategy. Unlike traditional methods that rely on harsh conditions or multi-step sequences, this protocol utilizes inexpensive and readily available aromatic carboxylic acids as direct arylation reagents. By integrating ligand exchange and decarboxylation mechanisms, the process forms aryl-palladium species in situ, which subsequently undergo nucleophilic addition to cyano groups followed by ring closure. This approach not only enhances atom economy but also streamlines the supply chain for high-purity pharmaceutical intermediates by removing the dependency on pre-functionalized halides.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of 2,5-disubstituted imidazole skeletons has been plagued by significant synthetic inefficiencies that hinder large-scale manufacturing. Conventional strategies typically necessitate the preparation of highly activated reaction substrates through lengthy multi-step sequences, often involving toxic reagents and protecting group manipulations that drastically reduce overall yield. Alternatively, post-synthetic modification of existing imidazole rings via classic cross-coupling reactions requires the use of pre-functionalized aryl halides or boronic acids, which are substantially more expensive and less stable than their carboxylic acid counterparts. Furthermore, these traditional pathways frequently demand harsh reaction conditions, such as strong bases or elevated temperatures that compromise functional group tolerance, leading to complex impurity profiles that are difficult to purge during purification. The cumulative effect of these limitations is a high cost of goods and extended lead times, creating bottlenecks for procurement teams seeking reliable sources of complex heterocyclic building blocks for drug development pipelines.
The Novel Approach
The methodology disclosed in CN113582929B represents a paradigm shift by enabling the direct use of simple 2,6-dimethoxybenzoic acid as the arylation source in a one-pot tandem reaction. This novel approach bypasses the need for pre-activation of the aromatic ring, relying instead on a sophisticated palladium catalytic cycle that activates the carboxylic acid through decarboxylation. The reaction proceeds under relatively mild thermal conditions at 120°C in 1,4-dioxane, utilizing a specific ligand system comprising 2,9-dimethyl-1,10-phenanthroline to stabilize the active catalytic species. Crucially, the process operates without the need for external metal oxidants, which simplifies the reaction matrix and reduces the environmental footprint associated with heavy metal waste. This streamlined protocol allows for the direct convergence of cyano-substituted amides and benzoic acid derivatives, significantly shortening the synthetic route and improving the overall step economy. For supply chain managers, this translates to a more robust manufacturing process with fewer unit operations and reduced exposure to volatile raw material markets associated with specialized coupling partners.
Mechanistic Insights into Pd-Catalyzed Decarboxylative Cyclization
The mechanistic underpinning of this transformation involves a carefully orchestrated sequence of organometallic steps that ensure high chemoselectivity and conversion efficiency. The cycle initiates with the coordination of the palladium catalyst, specifically Pd(TFA)2, to the carboxylate oxygen of the 2,6-dimethoxybenzoic acid substrate, facilitated by the bidentate nitrogen ligand. Following ligand exchange, the system undergoes a thermal decarboxylation event, releasing carbon dioxide and generating a reactive aryl-palladium intermediate. This electrophilic metal species then engages in a nucleophilic addition across the nitrile triple bond of the cyano-substituted amide, forming a new carbon-carbon bond and an imino-palladium species. The presence of heptafluorobutyric acid (HFBA) as an additive plays a critical role in protonating intermediates and facilitating the subsequent intramolecular cyclization. This tandem sequence culminates in the formation of the imidazole ring with the elimination of the palladium catalyst, which re-enters the cycle. The elegance of this mechanism lies in its ability to construct two new bonds and a heterocyclic ring in a single operational step, minimizing the formation of side products that typically arise from discrete stepwise syntheses.
From an impurity control perspective, the mildness of the reaction conditions and the specificity of the decarboxylative pathway contribute to a cleaner crude reaction profile. Traditional coupling methods often suffer from homocoupling of aryl halides or protodeboronation side reactions, which generate structurally similar impurities that are notoriously difficult to separate via crystallization or chromatography. In contrast, the decarboxylative route described here avoids these specific degradation pathways, as the carboxylic acid starting material is less prone to oxidative homocoupling under the nitrogen atmosphere employed. Furthermore, the use of a defined ligand system helps suppress non-productive catalyst decomposition, ensuring that the majority of the palladium species remain active for the desired transformation. This results in a final product with a simplified impurity spectrum, reducing the burden on quality control laboratories and allowing for more efficient purification strategies. For R&D directors, this level of chemical fidelity is paramount when scaling up processes for clinical trial materials where strict purity specifications must be met.
How to Synthesize 2,5-Disubstituted Imidazoles Efficiently
The practical execution of this synthesis is designed for reproducibility and ease of handling in standard laboratory or pilot plant settings. The protocol involves charging a reaction vessel with the cyano-substituted amide substrate, 2,6-dimethoxybenzoic acid, the palladium catalyst, and the phenanthroline ligand in 1,4-dioxane solvent. The addition of heptafluorobutyric acid is critical for promoting the cyclization step and maintaining the acidity required for catalyst turnover. The detailed standardized synthesis steps are provided in the guide below to ensure consistent results across different batches.
- Combine cyano-substituted amide, 2,6-dimethoxybenzoic acid, Pd(TFA)2 catalyst, and phenanthroline ligand in 1,4-dioxane solvent.
- Add heptafluorobutyric acid (HFBA) as an additive and heat the mixture to 120°C under a nitrogen atmosphere for 12 hours.
- Upon completion, cool the reaction, perform aqueous workup with sodium carbonate and brine, dry the organic phase, and purify via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement professionals and supply chain strategists, the adoption of this patented synthetic route offers tangible benefits that extend beyond mere chemical novelty. The primary advantage lies in the substitution of expensive, specialized coupling partners with commodity chemicals. Aromatic carboxylic acids are produced on a massive industrial scale for various applications, making them significantly cheaper and more readily available than aryl boronic acids or iodides, which often suffer from supply volatility and price fluctuations. By shifting the raw material base to these stable acids, manufacturers can achieve substantial cost savings in pharmaceutical intermediate manufacturing without compromising on yield or quality. Additionally, the elimination of external metal oxidants reduces the consumption of auxiliary reagents, further driving down the variable costs associated with each production batch. This economic efficiency is compounded by the high atom economy of the reaction, where the majority of the starting material mass is incorporated into the final product, minimizing waste disposal costs.
Enhanced supply chain reliability is another critical benefit derived from this methodology. The robustness of the reaction conditions, operating at 120°C under nitrogen, allows for scalability from gram-level experiments to multi-kilogram production runs without significant re-optimization. The broad substrate scope demonstrated in the patent, accommodating diverse R1 groups including alkyl, aryl, and heteroaryl moieties, means that a single platform technology can be applied to synthesize a wide library of analogues. This flexibility reduces the need for developing unique, bespoke processes for each new candidate molecule, thereby accelerating the timeline from discovery to commercial supply. For supply chain heads, this translates to reduced lead times for high-purity pharmaceutical intermediates and a lower risk of production delays caused by complex process validation requirements. The ability to source raw materials from multiple global suppliers due to their commodity status further de-risks the supply chain against geopolitical or logistical disruptions.
Scalability and environmental compliance are increasingly important metrics for modern chemical manufacturing, and this process excels in both areas. The one-pot nature of the reaction reduces the number of isolation and purification steps, which in turn lowers solvent consumption and energy usage per kilogram of product. The absence of stoichiometric metal oxidants means that the waste stream contains lower levels of heavy metals, simplifying wastewater treatment and ensuring compliance with stringent environmental regulations. As the pharmaceutical industry moves towards greener chemistry principles, adopting such atom-economical and waste-reducing technologies provides a competitive advantage. The process has been shown to proceed smoothly to completion, indicating that it can be reliably scaled up to meet commercial demand. This combination of operational simplicity, cost efficiency, and environmental stewardship makes the technology highly attractive for contract development and manufacturing organizations looking to optimize their portfolio of synthetic capabilities.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthetic protocol. These insights are derived directly from the experimental data and beneficial effects outlined in the patent documentation, providing clarity on reaction parameters and scope.
Q: What are the key advantages of using carboxylic acids over aryl halides in this imidazole synthesis?
A: Using simple aromatic carboxylic acids eliminates the need for pre-functionalized and often expensive aryl halides or boronic acids. This decarboxylative approach improves atom economy, reduces hazardous waste generation, and utilizes cheaper, more stable starting materials, significantly lowering the overall cost of goods for pharmaceutical intermediates.
Q: Does this Pd-catalyzed method require external metal oxidants?
A: No, one of the distinct benefits of this patented protocol is that it proceeds smoothly without the addition of external metal oxidants. The reaction relies on the decarboxylation process to drive the formation of the aryl-palladium species, simplifying the reaction mixture and reducing the burden on downstream purification and heavy metal removal processes.
Q: What is the substrate scope for the R1 group in the cyano-substituted amide?
A: The method demonstrates excellent functional group tolerance. The R1 group can be varied widely to include alkyl groups (methyl, tert-butyl, cyclopropyl, cyclohexyl), aryl groups (phenyl, substituted phenyls with electron-withdrawing or donating groups), and heteroaryl groups (furan, thiophene, naphthyl), making it highly versatile for diverse drug discovery campaigns.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2,5-Disubstituted Imidazoles Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic value of advanced synthetic methodologies like the Pd-catalyzed decarboxylative cyclization in securing a competitive edge in the pharmaceutical market. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory discoveries are successfully translated into robust industrial processes. We are committed to delivering high-purity 2,5-disubstituted imidazoles that meet stringent purity specifications required for clinical and commercial applications. Our rigorous QC labs employ state-of-the-art analytical techniques to verify the identity and purity of every batch, guaranteeing consistency and reliability for our global partners. By leveraging our expertise in transition metal catalysis and heterocyclic chemistry, we can help you navigate the complexities of supply chain management and process optimization.
We invite you to collaborate with us to explore how this innovative synthesis can enhance your drug development pipeline. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific project needs. We are prepared to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities align with your quality and timeline requirements. Let us be your partner in transforming complex chemical challenges into commercially viable solutions.