Advanced Copper-Catalyzed Synthesis of 2-Sulfonamidopyridine Derivatives for Commercial Scale-Up
Advanced Copper-Catalyzed Synthesis of 2-Sulfonamidopyridine Derivatives for Commercial Scale-Up
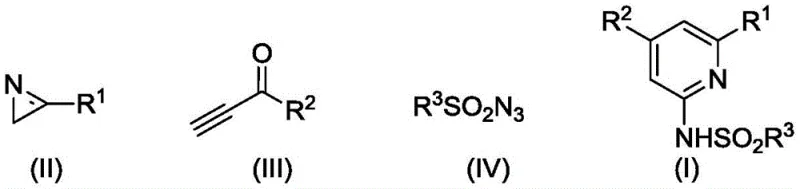
The pharmaceutical industry continuously seeks robust methodologies for constructing nitrogen-containing heterocycles, particularly the 2-aminopyridine scaffold, which serves as a critical pharmacophore in numerous bioactive molecules. A significant breakthrough in this domain is documented in Chinese Patent CN114315708B, which discloses a highly efficient preparation method for 2-sulfonamidopyridine derivatives. This innovative approach utilizes a tandem copper-catalyzed reaction sequence involving 3-aryl-2H-aziridines, terminal alkynyl ketones, and sulfonyl azides. Unlike traditional multi-step syntheses that often suffer from low atom economy and harsh conditions, this protocol achieves the target architecture in a streamlined one-pot fashion. The process leverages the synergistic effects of a copper catalyst and an amine ligand to drive a complex cascade of cycloaddition, ring-opening, and recyclization events. For R&D teams and procurement specialists alike, this technology represents a pivotal shift towards more sustainable and cost-effective manufacturing of high-purity pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of 2-aminopyridine cores has relied heavily on the Chichibabin reaction or modifications of pre-existing pyridine rings, such as the amination of 2-halopyridines. While these classical strategies have served the industry for decades, they are increasingly viewed as suboptimal for modern drug discovery and process chemistry. The Chichibabin reaction, for instance, typically requires extremely harsh conditions, including high temperatures and strong bases like sodium amide, which limits its compatibility with sensitive functional groups often present in advanced intermediates. Furthermore, metal-catalyzed coupling reactions on halogenated pyridines, while versatile, introduce additional synthetic steps and generate stoichiometric amounts of salt waste. Alternative strategies involving [5C+1N] cyclization or reactions with cyanamide derivatives often suffer from the availability of exotic starting materials and complicated purification protocols. These legacy methods frequently result in lower overall yields and higher production costs, creating bottlenecks in the supply chain for reliable pharmaceutical intermediate suppliers aiming to scale up production efficiently.
The Novel Approach
In stark contrast to these conventional limitations, the methodology described in CN114315708B offers a transformative solution by assembling the pyridine ring de novo from simple, commercially available precursors. This novel approach employs a copper-catalyzed cascade that seamlessly integrates a copper-catalyzed azide-alkyne cycloaddition (CuAAC) with subsequent ring-opening and cycloaddition steps. By reacting 3-aryl-2H-aziridines with terminal alkynyl ketones and sulfonyl azides, the process constructs the heterocyclic core with exceptional atom economy. The reaction conditions are notably mild, typically proceeding at moderate temperatures around 80°C in common solvents like acetonitrile. This operational simplicity eliminates the need for cryogenic conditions or specialized high-pressure equipment, thereby reducing the capital expenditure required for implementation. Moreover, the broad substrate scope allows for the introduction of diverse substituents, enabling the rapid generation of structural analogs essential for structure-activity relationship (SAR) studies without the need for extensive route redesign.
Mechanistic Insights into Copper-Catalyzed Cascade Cyclization
The elegance of this synthesis lies in its intricate mechanistic pathway, which orchestrates multiple bond-forming events in a single vessel. As illustrated in the detailed mechanism, the reaction initiates with the copper-catalyzed 1,3-dipolar cycloaddition between the sulfonyl azide and the terminal alkyne of the ynone. This forms a copper-triazole complex, which subsequently undergoes a ring-opening rearrangement to generate a reactive ketenimine intermediate. This electrophilic species is then intercepted by the nucleophilic nitrogen of the 3-aryl-2H-aziridine via a nucleophilic addition. Following this, an enol tautomerization facilitates an intramolecular [4+2] cycloaddition, leading to a fused bicyclic intermediate. Under the thermal conditions of the reaction, the strained aziridine ring opens due to ring tension, stabilizing as a 1,3-oxazine derivative. This intermediate is the precursor to the final product, setting the stage for the final aromatization step.
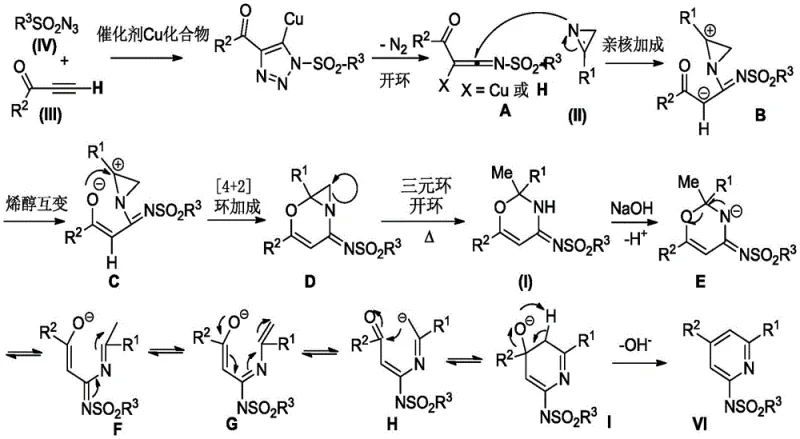
The final transformation involves the addition of a base, such as sodium hydroxide, which triggers a ring cleavage and recyclization process. This base-mediated step is crucial for driving the equilibrium towards the thermodynamically stable aromatic pyridine system. The mechanism proceeds through a series of proton transfers and bond reorganizations, effectively expelling the oxygen atom incorporated during the earlier oxazine formation and establishing the fully conjugated 2-sulfonamidopyridine skeleton. Understanding this mechanism is vital for process optimization, as it highlights the critical role of the copper catalyst in activating the azide and the necessity of the base for the final aromatization. Impurity control is inherently managed by the specificity of the copper-ligand complex, which minimizes side reactions such as homocoupling of the alkyne or non-selective aziridine ring opening. This high level of chemoselectivity ensures that the crude reaction mixture is relatively clean, simplifying downstream purification and enhancing the overall purity profile of the final API intermediate.
How to Synthesize 2-Sulfonamidopyridine Derivatives Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires careful attention to the stoichiometry and order of addition to maximize yield and safety. The standard protocol involves dissolving the 3-aryl-2H-aziridine, the terminal alkynyl ketone, and the sulfonyl azide in a polar aprotic solvent, with acetonitrile being the preferred medium due to its superior solvation properties and reaction kinetics. A catalytic amount of cuprous chloride and a stoichiometric amount of triethylamine are added to initiate the cascade. The detailed standardized synthesis steps are outlined in the guide below, ensuring reproducibility and safety compliance for technical teams looking to adopt this methodology.
- Mix 3-aryl-2H-aziridine, terminal ynone, sulfonyl azide, copper catalyst (e.g., CuCl), and amine ligand (e.g., TEA) in acetonitrile.
- Heat the mixture to 80°C for 6 hours to allow the CuAAC and ring-opening cascade to form the 1,3-oxazine intermediate.
- Add sodium hydroxide and continue heating for 4 hours to induce ring cleavage and recyclization into the final pyridine derivative.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented technology offers substantial benefits that directly address the pain points of procurement managers and supply chain directors. The shift from multi-step, protection-group-heavy syntheses to a direct one-pot assembly significantly reduces the number of unit operations required. This consolidation translates into a drastic simplification of the manufacturing workflow, lowering labor costs and minimizing the footprint required for production. Furthermore, the reliance on commodity chemicals such as copper chloride, triethylamine, and sodium hydroxide ensures that the raw material costs remain low and stable, insulating the supply chain from the volatility often associated with specialized organometallic reagents. The high atom economy of the reaction means that less raw material is wasted as byproduct, aligning with green chemistry principles and reducing waste disposal costs, which is a critical factor in total cost of ownership calculations.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts like palladium or rhodium, which are often required for cross-coupling alternatives, results in significant cost savings. Additionally, the use of inexpensive copper salts and the ability to run the reaction at moderate temperatures reduces energy consumption. The high yields reported, often exceeding 80%, mean that less starting material is needed to produce the same amount of product, further driving down the cost per kilogram. The simplified workup procedure, which avoids complex extractions or distillations, also contributes to lower operational expenditures.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis, including aryl aziridines, ynones, and sulfonyl azides, are readily available from global chemical suppliers, reducing the risk of supply disruptions. The robustness of the reaction conditions allows for flexibility in sourcing, as the process is tolerant to minor variations in reagent quality. This reliability ensures consistent production schedules and shorter lead times for high-purity pharmaceutical intermediates, enabling manufacturers to respond more agilely to market demands. The scalability of the process from gram to kilogram scale has been demonstrated, providing confidence for long-term supply agreements.
- Scalability and Environmental Compliance: The process generates minimal hazardous waste compared to traditional methods that utilize heavy metals or generate large volumes of saline waste. The solvents used, primarily acetonitrile, are easily recoverable and recyclable, supporting sustainability goals. The absence of highly toxic reagents simplifies regulatory compliance and reduces the burden on environmental health and safety teams. This environmental friendliness not only mitigates regulatory risk but also enhances the corporate social responsibility profile of the manufacturing entity, making it a more attractive partner for major pharmaceutical companies focused on sustainable sourcing.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthesis route. These insights are derived directly from the experimental data and optimization studies presented in the patent documentation, providing a reliable foundation for process development decisions. Understanding these nuances helps in troubleshooting potential issues and maximizing the efficiency of the production campaign.
Q: What is the optimal catalyst system for this synthesis?
A: According to patent data, cuprous chloride (CuCl) combined with triethylamine (TEA) as a ligand provides the highest yields, significantly outperforming divalent copper salts or ligand-free conditions.
Q: Can this method tolerate diverse functional groups?
A: Yes, the protocol demonstrates broad substrate scope, successfully accommodating electron-withdrawing groups like trifluoromethyl and halogens, as well as electron-donating methyl groups on the aryl rings.
Q: Is the purification process complex?
A: The workup is straightforward, involving standard extraction with ethyl acetate and brine, followed by silica gel chromatography, yielding products with purity exceeding 98%.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Sulfonamidopyridine Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic value of adopting cutting-edge synthetic methodologies like the one described in CN114315708B to enhance our portfolio of pharmaceutical intermediates. Our team of expert chemists has extensively evaluated this copper-catalyzed route and confirmed its viability for large-scale production. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from benchtop to plant floor is seamless and efficient. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 2-sulfonamidopyridine derivative we deliver meets the highest industry standards for quality and consistency.
We invite procurement leaders and R&D directors to collaborate with us to optimize their supply chains using this advanced technology. By leveraging our expertise, you can achieve significant cost reduction in API manufacturing while securing a stable supply of critical building blocks. We encourage you to contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific project needs. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our capabilities can support your drug development timelines and commercial manufacturing goals.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →