Advanced Rhodium-Catalyzed Synthesis of Chiral Cyclopropyl Pyrimidine Nucleoside Analogs for Antiviral Drug Development
The pharmaceutical landscape for antiviral therapeutics has been revolutionized by the advent of nucleoside analogs, particularly those featuring complex carbocyclic structures such as Sofosbuvir and Besifovir. A pivotal development in this field is disclosed in patent CN108314655B, which details a robust method for synthesizing three-membered carbocyclic pyrimidine nucleoside analogs via rhodium-catalyzed asymmetric cyclopropanation. This technology addresses a critical bottleneck in organic synthesis: the efficient construction of chiral quaternary carbon centers within a cyclopropane ring fused to a pyrimidine base. For R&D directors and procurement specialists seeking reliable pharmaceutical intermediates supplier partnerships, this patent represents a significant leap forward. The methodology transforms simple 1-vinyl substituted pyrimidines and aryl diazo esters into high-value chiral scaffolds with exceptional stereocontrol. By leveraging chiral rhodium catalysis, the process bypasses the tedious multi-step protections and deprotections typical of legacy routes, offering a direct pathway to compounds with potent anti-HBV and anti-HCV activity. This report analyzes the technical depth and commercial viability of this synthesis, highlighting its potential to streamline the supply chain for next-generation antiviral medications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of chiral three-membered carbocyclic nucleosides has been plagued by inefficiency and structural complexity. Traditional strategies often rely on a retrosynthetic disconnection that prioritizes the formation of the carbocyclic ring first, followed by the laborious introduction of the nitrogenous base. This approach typically necessitates multiple synthetic steps involving harsh conditions to install amino or ureido groups onto the carbocycle before finally constructing the purine or pyrimidine ring system. Such multi-step designs not only suffer from cumulative yield losses but also introduce significant challenges in controlling the absolute configuration of the chiral centers. Furthermore, the separation of diastereomers and enantiomers in these late-stage intermediates often requires expensive chiral resolution techniques, drastically inflating the cost of goods. For supply chain managers, these legacy processes represent a vulnerability, as long lead times and low overall throughput hinder the ability to respond rapidly to market demands for antiviral APIs.
The Novel Approach
In stark contrast, the novel approach described in the patent utilizes a convergent strategy that constructs the core scaffold in a single catalytic event. By reacting 1-vinyl substituted pyrimidine derivatives directly with aryl diazo esters in the presence of a chiral rhodium catalyst, the method installs the cyclopropane ring and the quaternary carbon center simultaneously. This transformation is remarkably efficient, converting simple, commercially available starting materials into complex nucleoside analogs with high fidelity. The reaction tolerates a wide variety of substituents on both the pyrimidine ring and the diazo component, allowing for the rapid generation of diverse libraries of analogs for structure-activity relationship (SAR) studies. This shift from a linear, multi-step synthesis to a concise, catalytic cycle fundamentally alters the economics of production, offering a pathway to cost reduction in pharmaceutical intermediates manufacturing by minimizing waste, solvent usage, and processing time.
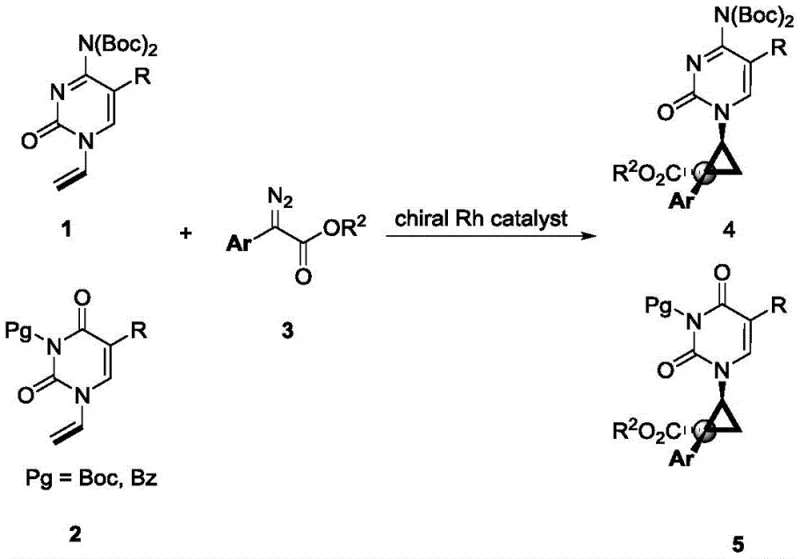
Mechanistic Insights into Rhodium-Catalyzed Asymmetric Cyclopropanation
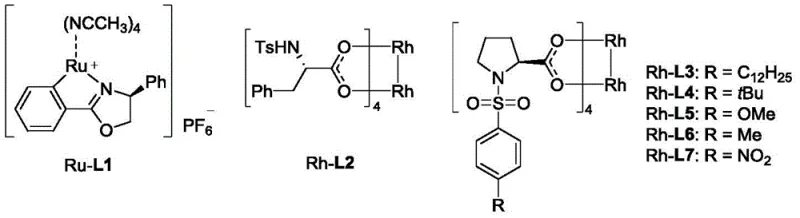
The success of this synthesis hinges on the precise engineering of the chiral rhodium catalyst system. The reaction proceeds through the formation of a reactive rhodium-carbenoid species generated from the decomposition of the diazo ester. This electrophilic metal-carbene intermediate then undergoes a concerted cyclopropanation with the electron-deficient double bond of the 1-vinyl pyrimidine substrate. The chirality of the final product is dictated by the chiral environment surrounding the rhodium center, which directs the facial selectivity of the alkene approach. Extensive screening identified that dirhodium tetracarboxylate catalysts bearing specific chiral ligands, particularly the Rh-L7 variant with a nitro-substituted phenyl group, provide superior stereoinduction. The steric bulk and electronic properties of the ligand create a rigid chiral pocket that effectively discriminates between the prochiral faces of the vinyl group, ensuring the formation of the desired (1'S, 2'R) or equivalent configuration with high enantiomeric excess.
Impurity control is another critical aspect where this mechanistic understanding pays dividends. The use of protected pyrimidine substrates (with Boc or Bz groups) is essential to prevent competitive N-H insertion reactions, which would otherwise lead to intractable mixtures of byproducts. By carefully selecting the protecting group and optimizing the reaction temperature to -50°C, the process suppresses non-selective background reactions and favors the desired cyclopropanation pathway. The result is a crude reaction profile that is exceptionally clean, simplifying downstream purification and ensuring that the final isolated product meets stringent purity specifications required for GMP manufacturing. This level of control over the reaction trajectory is vital for maintaining consistent quality in the production of high-purity nucleoside analogs.
How to Synthesize Chiral Cyclopropyl Pyrimidine Nucleoside Analogs Efficiently
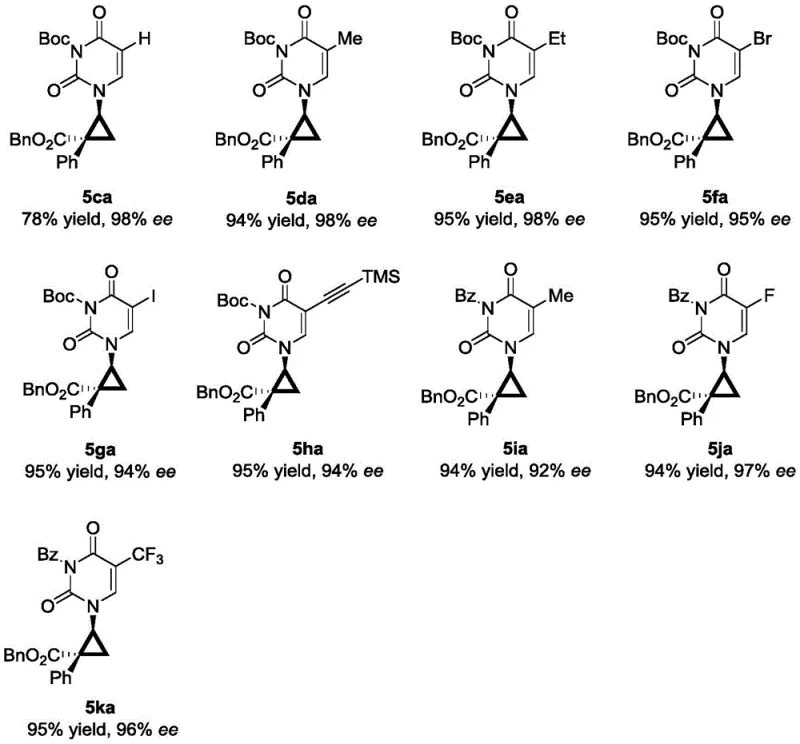
The operational simplicity of this rhodium-catalyzed protocol makes it highly attractive for process chemistry teams looking to implement new routes quickly. The standard procedure involves dissolving the 1-vinyl pyrimidine substrate and the catalyst in a dry solvent like toluene, followed by cooling the mixture to cryogenic temperatures. The diazo ester, dissolved in a separate portion of solvent, is then added slowly via a syringe pump to maintain a low steady-state concentration of the reactive carbenoid species, which further enhances selectivity. While the general concept is straightforward, precise control over stoichiometry, addition rate, and temperature is paramount to achieving the reported yields of 75-96% and ee values up to 99%. For detailed operational parameters and safety considerations regarding diazo compounds, please refer to the standardized guide below.
- Dissolve 1-vinyl substituted pyrimidine and the chiral rhodium catalyst (preferably Rh-L7) in an appropriate solvent such as toluene.
- Cool the reaction mixture to a temperature between -20°C and -60°C, optimally around -50°C, to ensure high enantioselectivity.
- Slowly add the aryl diazo ester solution dropwise to the reaction mixture over several hours, then purify the resulting chiral cyclopropane product via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this technology offers compelling advantages that extend beyond mere chemical elegance. For procurement managers, the reliance on readily available starting materials such as vinyl pyrimidines and substituted phenylacetic acid derivatives ensures a stable and resilient supply chain. Unlike exotic reagents that may face sourcing bottlenecks, these precursors are commodity chemicals produced at scale, mitigating the risk of supply disruption. Furthermore, the high atom economy of the cyclopropanation reaction means that a significant proportion of the reactant mass is incorporated into the final product, reducing the burden of waste disposal and lowering the overall environmental footprint of the manufacturing process.
- Cost Reduction in Manufacturing: The streamlined nature of this synthesis eliminates the need for multiple isolation and purification steps associated with traditional linear routes. By consolidating the formation of the carbocycle and the installation of the chiral center into a single catalytic step, manufacturers can significantly reduce labor costs, solvent consumption, and equipment occupancy time. The high yields and excellent stereoselectivity minimize the loss of valuable intermediates, directly contributing to a lower cost per kilogram of the final API intermediate. Additionally, the ability to use robust catalysts that function effectively at low loadings helps to offset the cost of the precious metal, making the process economically viable for large-scale production.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions contributes to greater predictability in production scheduling. The tolerance of the catalyst system to various functional groups allows for flexibility in raw material sourcing, as different substituted diazo esters can be used without requiring a complete re-optimization of the process. This flexibility is crucial for maintaining continuity of supply in the face of fluctuating raw material markets. Moreover, the simplified workflow reduces the number of potential failure points in the manufacturing line, ensuring that delivery timelines for critical antiviral intermediates can be met consistently.
- Scalability and Environmental Compliance: Scaling this reaction from gram to kilogram quantities is facilitated by the use of standard organic solvents like toluene and manageable temperature ranges. The exothermic nature of diazo decomposition is safely controlled through slow addition protocols, a technique that translates well to industrial reactors. From an environmental standpoint, the reduction in step count inherently lowers the E-factor (mass of waste per mass of product), aligning with modern green chemistry principles. This not only simplifies regulatory compliance regarding waste discharge but also enhances the sustainability profile of the final pharmaceutical product, a key consideration for major global pharma partners.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this rhodium-catalyzed synthesis. These insights are derived directly from the experimental data and optimization studies presented in the patent literature, providing a clear picture of the technology's capabilities and limitations for potential adopters.
Q: What is the primary advantage of this rhodium-catalyzed method over traditional synthesis?
A: This method directly constructs the chiral quaternary carbon center in a single step using 1-vinyl substituted pyrimidine and aryl diazo ester, avoiding the complex multi-step sequences required by conventional methods that build the base onto a pre-formed carbocycle.
Q: What level of enantioselectivity can be achieved with this process?
A: By utilizing the optimized chiral rhodium catalyst Rh-L7 at low temperatures (-50°C), the process consistently achieves excellent enantioselectivity, with ee values reaching up to 99% across various substrates.
Q: Is this synthesis method suitable for large-scale production?
A: Yes, the reaction utilizes readily available raw materials and operates under manageable conditions (toluene solvent, -50°C), making it highly amenable to commercial scale-up for the production of antiviral drug intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral Cyclopropyl Pyrimidine Nucleoside Analogs Supplier
The transition from laboratory discovery to commercial reality requires a partner with deep technical expertise and proven manufacturing capabilities. NINGBO INNO PHARMCHEM stands at the forefront of custom synthesis, possessing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facility is equipped with state-of-the-art cryogenic reactors capable of maintaining the precise -50°C conditions required for this high-selectivity cyclopropanation, ensuring that the exceptional enantiomeric purity demonstrated in the lab is preserved at scale. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of nucleoside intermediate meets the exacting standards of the global pharmaceutical industry.
We invite R&D and procurement leaders to collaborate with us to evaluate the feasibility of this route for your specific pipeline projects. Our technical team is prepared to provide a Customized Cost-Saving Analysis tailored to your volume requirements, demonstrating how this catalytic method can optimize your COGS. Please contact our technical procurement team today to request specific COA data for our catalog intermediates or to discuss route feasibility assessments for your proprietary targets. Let us help you accelerate the development of life-saving antiviral therapies through superior chemical manufacturing.