Advanced Two-Step Synthesis of Abemaciclib Intermediate for Commercial Scale-Up
Introduction to Advanced CDK4/6 Inhibitor Intermediate Synthesis
The global pharmaceutical landscape has witnessed a paradigm shift in the treatment of hormone receptor-positive breast cancer, driven largely by the advent of Cyclin-Dependent Kinase 4 and 6 (CDK4/6) inhibitors. Among these revolutionary therapeutics, Abemaciclib stands out as a potent oral agent that has significantly improved patient outcomes by targeting cell cycle pathways. However, the commercial viability of such life-saving medications hinges critically on the efficiency, safety, and scalability of their synthetic intermediates. A pivotal breakthrough in this domain is detailed in patent CN112028834B, which discloses a novel, streamlined synthesis method for 6-bromo-4-fluoro-1-isopropyl-2-methyl-1H-benzo[d]imidazole, a key building block for Abemaciclib. This technical insight report analyzes the proprietary methodology outlined in the patent, highlighting how it overcomes historical manufacturing bottlenecks through a concise two-step sequence involving condensation and reductive alkylation. For R&D directors and supply chain leaders, understanding this process is essential for securing a reliable supply of high-purity pharmaceutical intermediates while mitigating the operational risks associated with traditional hazardous chemistry.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies, such as those disclosed in US20100160340, have long plagued the industry with complex multi-step sequences that introduce significant safety and environmental liabilities. These conventional routes typically rely on the preparation of N-isopropylacetamide using acetyl chloride, a reagent known for its extreme instability in air, tendency to smoke, and high flammability, creating substantial storage and handling hazards during industrial transfer. Furthermore, the subsequent cyclization steps often necessitate the use of phosphorus oxychloride and strong bases like potassium tert-butoxide or sodium hydride. The use of sodium hydride is particularly concerning due to its pyrophoric nature, posing a severe risk of spontaneous combustion upon contact with moisture, which is unacceptable for large-scale commercial production. Additionally, the harsh basic conditions employed in these legacy processes frequently lead to unwanted nucleophilic aromatic substitution, where the critical fluorine atom on the benzene ring is displaced by tert-butoxy groups, generating difficult-to-remove impurities that compromise the quality of the final API.
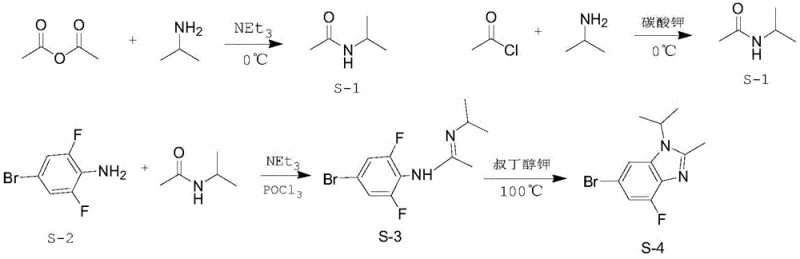
The Novel Approach
In stark contrast to these cumbersome legacy protocols, the innovative strategy presented in patent CN112028834B simplifies the entire manufacturing workflow into a highly efficient two-step process that prioritizes safety and atom economy. The new route initiates with a direct condensation reaction between 5-bromo-3-fluoro-1,2-phenylenediamine and acetic acid, bypassing the need for dangerous acyl chlorides entirely. This is followed by a mild reductive alkylation using acetone and a selective reducing agent, such as sodium triacetoxyborohydride, to install the isopropyl group at the N1 position. By operating at moderate temperatures ranging from 0°C to 80°C and utilizing common solvents like toluene and n-heptane, this method drastically reduces the potential for thermal runaway reactions. The elimination of strong bases ensures that the sensitive fluorine substituent remains intact, thereby preserving the structural integrity required for downstream biological activity and significantly simplifying the purification profile.
Mechanistic Insights into Reductive Alkylation and Cyclization
The core chemical transformation in this novel synthesis relies on a sophisticated interplay between acid-catalyzed condensation and hydride-mediated reduction, mechanisms that are fundamental to achieving high selectivity without compromising functional group tolerance. In the first stage, the diamine substrate undergoes a dehydration condensation with acetic acid, likely proceeding through an amide intermediate that subsequently cyclizes to form the benzimidazole core. This step is thermodynamically driven by the removal of water and is optimized in high-boiling solvents like toluene at reflux temperatures around 110°C, ensuring complete conversion of the starting material while minimizing the formation of oligomeric byproducts. The second stage involves the activation of the benzimidazole nitrogen through the formation of an iminium ion intermediate upon reaction with acetone, which is then selectively reduced by the borohydride species. Unlike harsh alkylation methods that utilize alkyl halides and strong bases, this reductive amination pathway operates under near-neutral conditions, preventing the deprotonation of the aromatic ring that leads to nucleophilic attack on the fluorine position.
Impurity control is inherently built into this mechanistic design, as the choice of sodium triacetoxyborohydride as the reducing agent offers superior chemoselectivity compared to lithium aluminum hydride or sodium borohydride in acidic media. This specificity ensures that the reduction targets only the transient iminium species generated in situ, leaving the aryl bromide and aryl fluoride moieties untouched. Furthermore, the workup procedure described in the patent leverages pH-controlled extractions using dilute sodium carbonate solutions to remove acidic residues and unreacted amines, followed by a strategic recrystallization step. The use of n-heptane as an anti-solvent facilitates the precipitation of the target molecule while keeping polar impurities and residual salts in the mother liquor. This robust purification capability allows manufacturers to consistently achieve purity levels exceeding 99%, a critical specification for regulatory compliance in the production of oncology drug substances.
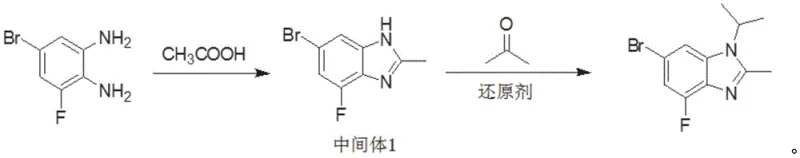
How to Synthesize 6-bromo-4-fluoro-1-isopropyl-2-methyl-1H-benzo[d]imidazole Efficiently
Implementing this synthesis protocol requires precise control over reaction parameters to maximize yield and ensure operator safety, leveraging the detailed embodiments provided in the patent documentation. The process begins with the charging of 5-bromo-3-fluoro-1,2-phenylenediamine into a reactor equipped with efficient agitation and temperature control, followed by the addition of toluene and acetic acid to initiate the cyclization. Once the intermediate benzimidazole is formed and isolated via aqueous workup, it is subjected to reductive alkylation conditions where stoichiometry and addition rates of the reducing agent are critical to prevent exotherms. The detailed standardized synthesis steps, including specific molar ratios, addition times, and crystallization cooling profiles, are outlined below to guide process engineers in replicating this high-efficiency route.
- Condense 5-bromo-3-fluoro-1,2-phenylenediamine with acetic acid in toluene at reflux (110°C) to form the benzimidazole core.
- Perform reductive alkylation by reacting the intermediate with acetone and sodium triacetoxyborohydride at 50°C.
- Purify the crude product via aqueous washing and recrystallization using n-heptane to achieve 99% purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis route translates directly into tangible operational improvements and risk mitigation strategies that enhance the overall resilience of the pharmaceutical supply network. By transitioning away from hazardous reagents like sodium hydride and acetyl chloride, manufacturing facilities can significantly lower their insurance premiums and reduce the capital expenditure required for specialized explosion-proof infrastructure and waste neutralization systems. The simplified two-step sequence also shortens the production cycle time, allowing for faster turnover of batches and improved responsiveness to fluctuating market demands for CDK4/6 inhibitor intermediates. Furthermore, the use of recoverable solvents like toluene and n-heptane aligns with green chemistry principles, reducing the volume of hazardous waste disposal and lowering the environmental compliance burden on the production site.
- Cost Reduction in Manufacturing: The elimination of expensive and hazardous reagents such as phosphorus oxychloride and specialized alkylating agents results in a substantial decrease in raw material costs per kilogram of finished intermediate. Additionally, the high yield of over 80% reported in the patent examples means that less starting material is wasted, directly improving the cost-of-goods-sold (COGS) metrics for the final API. The simplified purification process, which avoids complex chromatography or multiple distillation steps, further reduces utility consumption and labor hours, driving down the overall manufacturing overhead.
- Enhanced Supply Chain Reliability: Sourcing stability is greatly improved because the key raw materials, including 5-bromo-3-fluoro-1,2-phenylenediamine and acetic acid, are commodity chemicals available from multiple global suppliers, reducing dependency on single-source vendors. The robustness of the reaction conditions, which tolerate minor variations in temperature and mixing without forming significant impurities, ensures consistent batch-to-batch quality, minimizing the risk of production delays caused by out-of-specification results. This reliability is crucial for maintaining continuous supply lines to downstream API manufacturers who operate on tight just-in-time schedules.
- Scalability and Environmental Compliance: The process is inherently scalable from laboratory benchtop to multi-ton commercial production due to the absence of extremely exothermic or gas-evolving steps that are difficult to manage in large reactors. The reduction in wastewater COD and ammonia nitrogen levels, achieved by avoiding dimethyl sulfoxide and other hard-to-treat solvents, simplifies effluent treatment and ensures compliance with increasingly stringent environmental regulations. This environmental compatibility not only protects the manufacturer from regulatory fines but also enhances the corporate sustainability profile, which is increasingly important for partnerships with major multinational pharmaceutical companies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology, providing clarity on its practical application and benefits for potential partners. These answers are derived directly from the experimental data and comparative analysis presented in the patent literature, ensuring accuracy and relevance for technical decision-makers evaluating this route for adoption.
Q: What is the overall yield and purity of the new synthesis method?
A: According to patent CN112028834B, the optimized two-step process achieves a total yield of over 80% and a final product purity of 99% after simple recrystallization.
Q: Why is this method safer than conventional routes using sodium hydride?
A: This method eliminates the use of pyrophoric sodium hydride and unstable acetyl chloride, replacing them with safer reagents like acetic acid and sodium triacetoxyborohydride, significantly reducing explosion risks.
Q: How does this route prevent fluorine substitution byproducts?
A: By avoiding strong bases like potassium tert-butoxide which can nucleophilically attack the fluorine position, this mild reductive alkylation pathway preserves the critical fluoro-substituent on the benzene ring.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 6-bromo-4-fluoro-1-isopropyl-2-methyl-1H-benzo[d]imidazole Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of next-generation oncology therapies depends on the availability of high-quality intermediates produced via safe and scalable processes. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive a consistent supply of material that meets rigorous quality standards. Our state-of-the-art facilities are equipped to handle the specific solvent systems and reaction conditions required for this reductive alkylation process, and our stringent purity specifications are enforced through rigorous QC labs utilizing advanced analytical techniques to guarantee every batch exceeds 99% purity.
We invite pharmaceutical innovators and procurement leaders to collaborate with us to leverage this advanced synthesis technology for their Abemaciclib supply chains. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements, as well as obtain specific COA data and route feasibility assessments to validate the integration of this intermediate into your manufacturing portfolio. Contact us today to discuss how we can support your clinical and commercial needs with reliable, cost-effective, and compliant chemical solutions.