Advanced Manufacturing of Progesterone Pyrazinamide Compounds for Oncology Applications
Advanced Manufacturing of Progesterone Pyrazinamide Compounds for Oncology Applications
The global demand for novel oncology therapeutics continues to escalate, driven by the urgent need for treatments with new mechanisms of action and reduced toxicity profiles. Patent CN110903341B introduces a groundbreaking class of progesterone pyrazinamide compounds, specifically engineered to target malignant tumors such as prostate, breast, and liver cancer. This technology represents a significant leap forward in steroid-based drug design, merging the structural integrity of the progesterone skeleton with the pharmacophoric potential of a pyrazine-amide moiety. For pharmaceutical developers and procurement specialists, this patent outlines a highly efficient, convergent synthesis strategy that bypasses many of the traditional bottlenecks associated with complex steroid functionalization. By leveraging a modular approach that constructs the molecule from two distinct precursors, the process offers a robust pathway for the commercial scale-up of complex pharmaceutical intermediates, ensuring a reliable supply chain for next-generation anticancer agents.
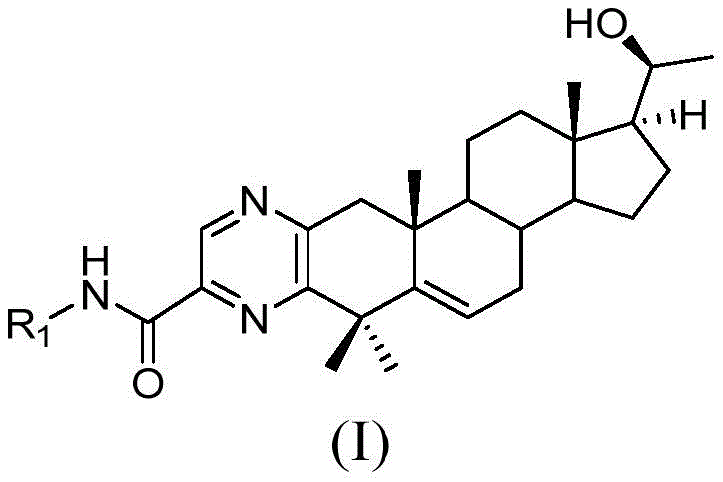
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the modification of the steroid nucleus to introduce nitrogen-containing heterocycles has been fraught with synthetic challenges. Traditional routes often involve direct functionalization of the steroid A-ring, which is sterically hindered and chemically sensitive. Introducing bulky aniline groups directly onto the steroid skeleton typically requires harsh reaction conditions, leading to poor regioselectivity and the formation of difficult-to-separate impurities. Furthermore, conventional methods frequently rely on multiple chromatographic purification steps after every transformation to remove side products and unreacted starting materials. This reliance on column chromatography is a major impediment to industrial scalability, as it drastically increases processing time, solvent waste, and overall production costs. The low atom economy and the necessity for protecting group manipulations that are difficult to remove cleanly further exacerbate the inefficiency of these legacy processes, making them unsuitable for the cost-sensitive manufacturing of high-volume API intermediates.
The Novel Approach
The methodology disclosed in CN110903341B fundamentally reimagines the construction of these hybrid molecules through a convergent synthesis strategy. Instead of struggling to build the heterocycle directly on the fragile steroid core, the inventors have devised a route where the pyrazine ring is formed by the condensation of two pre-functionalized fragments: a diamino acid derivative and a specific 2,3-ortho-dicarbonyl steroid intermediate. This approach allows for the optimization of each fragment independently under ideal conditions before the final coupling. Crucially, the patent highlights that the synthesis of these individual fragments can be achieved without the need for chromatographic column separation, relying instead on crystallization or simple extraction workups. This dramatic simplification of the purification protocol translates directly into higher throughput and significantly reduced operational expenditures. The final assembly involves a straightforward condensation followed by a selective reduction, yielding the target molecules with high purity and excellent yields, thereby establishing a superior manufacturing paradigm for high-purity pharmaceutical intermediates.
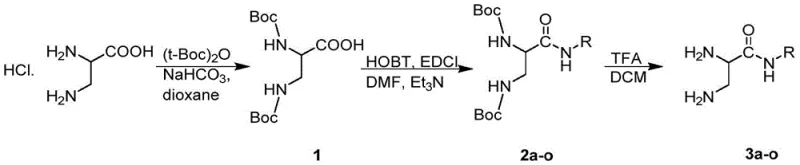
Mechanistic Insights into Convergent Fragment Coupling and Pyrazine Formation
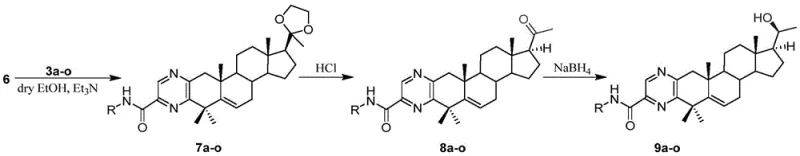
The chemical elegance of this synthesis lies in the precise orchestration of protecting group chemistry and condensation reactions. The first phase involves the preparation of the amino acid fragment (3a-o). Starting from DL-diaminopropionic acid hydrochloride, the amine functionalities are protected using di-tert-butyl dicarbonate ((t-Boc)2O) to prevent unwanted side reactions. Subsequent activation of the carboxylic acid using coupling reagents like EDCI and HOBt allows for the efficient formation of the amide bond with various substituted anilines or amines. The final deprotection step using trifluoroacetic acid (TFA) reveals the reactive diamino species required for the next stage. Parallel to this, the steroid fragment undergoes a sophisticated transformation sequence. The C-20 ketone is first protected as a ketal to prevent interference, followed by the introduction of gem-dimethyl groups at the C-4 position to modulate steric and electronic properties. The critical step is the oxidation at C-2 to generate the 2,3-dicarbonyl system, which serves as the electrophilic partner for the pyrazine ring closure.
The final convergence occurs when the nucleophilic diamino fragment attacks the electrophilic ortho-dicarbonyl steroid intermediate. This condensation spontaneously cyclizes to form the pyrazine ring, fusing the two distinct molecular architectures into a single bioactive entity. The mechanism proceeds through the formation of di-imine intermediates which then aromatize to stabilize the pyrazine core. Following this, the C-20 ketal protecting group is removed, and the resulting ketone is selectively reduced to the corresponding alcohol using sodium borohydride. This reduction is highly chemoselective, leaving the newly formed pyrazine ring and the steroid double bond intact. The entire sequence demonstrates exceptional control over stereochemistry and functional group tolerance, ensuring that the final product profile is clean and consistent. This mechanistic robustness is vital for maintaining stringent purity specifications required for clinical-grade materials.
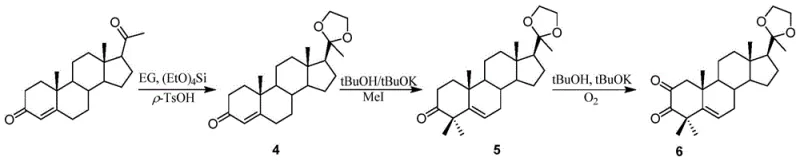
How to Synthesize Progesterone Pyrazinamide Efficiently
The synthesis of these potent anticancer intermediates is designed for operational simplicity and scalability. The process begins with the parallel preparation of the amino acid side chain and the modified steroid core, allowing for flexible inventory management of precursors. The coupling reaction is performed in anhydrous ethanol with catalytic acid, promoting rapid ring closure. Subsequent deprotection and reduction steps utilize common, inexpensive reagents like hydrochloric acid and sodium borohydride, avoiding the need for precious metal catalysts or specialized high-pressure reactors. The detailed standardized synthesis steps see the guide below for specific reaction parameters and workup procedures.
- Synthesize the amino acid molecular fragment (3a-o) via Boc protection, amide formation with substituted anilines, and subsequent deprotection.
- Prepare the key steroid intermediate (6) containing a 2,3-ortho-dicarbonyl system through C-20 ketal protection, C-4 dimethylation, and C-2 oxidation.
- Construct the final pyrazine ring by condensing the diamino fragment with the steroid ortho-dicarbonyl intermediate, followed by selective reduction of the C-20 ketone.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the technical features of this patent translate into tangible commercial benefits that directly impact the bottom line. The elimination of chromatographic purification during the intermediate stages is perhaps the most significant cost driver, as it removes a major bottleneck in production capacity and drastically reduces solvent procurement and disposal costs. The use of readily available starting materials like progesterone and DL-diaminopropionic acid ensures that the supply chain is not dependent on exotic or single-source reagents, thereby enhancing supply continuity and reducing lead time for high-purity pharmaceutical intermediates. Furthermore, the high yields reported in the examples, particularly in the final reduction step where yields exceed 90%, indicate a process with excellent material efficiency, minimizing waste and maximizing output per batch.
- Cost Reduction in Manufacturing: The process architecture inherently lowers production costs by removing the need for expensive silica gel column chromatography, which is both labor-intensive and solvent-heavy. By relying on crystallization and extraction for purification, the manufacturing footprint is reduced, and the consumption of hazardous organic solvents is minimized. Additionally, the avoidance of transition metal catalysts eliminates the costly and time-consuming step of heavy metal scavenging, which is often required to meet regulatory limits for residual metals in APIs. These factors combine to create a leaner, more cost-effective manufacturing process that offers substantial cost savings compared to traditional steroid modification routes.
- Enhanced Supply Chain Reliability: The synthetic route utilizes commodity chemicals and robust reaction conditions that are less susceptible to supply chain disruptions. Reagents such as Boc-anhydride, EDCI, and sodium borohydride are globally available from multiple suppliers, mitigating the risk of raw material shortages. The modular nature of the synthesis allows for the stockpiling of key intermediates (Fragment A and Fragment B), providing a buffer against demand fluctuations and ensuring consistent delivery schedules. This resilience is critical for maintaining the continuity of drug development pipelines and preventing costly delays in clinical trial material production.
- Scalability and Environmental Compliance: The chemistry described is inherently scalable, moving seamlessly from gram-scale laboratory synthesis to multi-kilogram pilot production without significant re-optimization. The reaction conditions, primarily involving reflux in common solvents like ethanol and dichloromethane, are well-suited for standard glass-lined or stainless steel reactors found in most GMP facilities. Moreover, the reduction in solvent usage and the absence of toxic heavy metals align with modern green chemistry principles, simplifying environmental compliance and waste treatment protocols. This facilitates a smoother regulatory approval process and supports corporate sustainability goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these progesterone pyrazinamide derivatives. These insights are derived directly from the experimental data and process descriptions within the patent documentation, providing clarity on the feasibility and performance of the technology.
Q: What represents the primary advantage of this synthesis route over traditional steroid modifications?
A: The primary advantage is the avoidance of chromatographic column separation during the synthesis of the two key molecular fragments. This significantly streamlines the process, reduces solvent consumption, and enhances overall throughput compared to traditional routes that often require extensive purification after each steroid functionalization step.
Q: Which specific cancer cell lines have shown sensitivity to these progesterone pyrazinamide derivatives?
A: According to the patent data, these compounds exhibit inhibitory activity against breast cancer (MCF-7), liver cancer (HepG2), and prostate cancer (PC-3) cell lines. Notably, the activity against prostate cancer cells is particularly significant, often surpassing the positive control 5-fluorouracil in specific structural variants.
Q: Is the final reduction step compatible with large-scale manufacturing?
A: Yes, the final reduction of the C-20 carbonyl group utilizes sodium borohydride (NaBH4) under mild conditions (ice-bath, methanol/THF). This is a standard, scalable reduction method widely used in industrial pharmaceutical manufacturing, ensuring high yields (up to 96%) without requiring exotic catalysts or high-pressure equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Progesterone Pyrazinamide Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the chemistry described in CN110903341B for the development of next-generation oncology therapeutics. As a premier CDMO partner, we possess the technical expertise and infrastructure to translate this innovative academic research into commercial reality. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can move smoothly from preclinical development to full-scale manufacturing. We are committed to delivering materials that meet stringent purity specifications, utilizing our rigorous QC labs to verify identity, potency, and impurity profiles at every stage of the synthesis.
We invite you to collaborate with us to leverage this advanced synthetic route for your drug discovery programs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how our optimized process can reduce your overall COGS. Please contact us today to request specific COA data for our reference standards and to discuss route feasibility assessments for your target analogues. Let us be your strategic partner in bringing these promising anticancer candidates to the market efficiently and reliably.