Scalable Synthesis of 7-Hydroxy-[1,2,4]triazolo[1,5-a]pyridine for Commercial Tucatinib Production
Scalable Synthesis of 7-Hydroxy-[1,2,4]triazolo[1,5-a]pyridine for Commercial Tucatinib Production
The pharmaceutical landscape for HER2-positive breast cancer treatment has been significantly advanced by the development of Tucatinib, a potent tyrosine kinase inhibitor. Central to the efficient manufacturing of this life-saving medication is the availability of high-quality key intermediates, specifically 7-hydroxy-[1,2,4]triazolo[1,5-a]pyridine. A groundbreaking preparation method disclosed in patent CN114671867A offers a transformative approach to synthesizing this critical building block. This technical insight report analyzes the novel synthetic pathway, contrasting it with legacy methods to highlight its potential for revolutionizing supply chain stability and cost structures for global API manufacturers. The strategic implementation of this chemistry allows for the direct construction of the triazolopyridine core without the burden of orthogonal protecting groups.
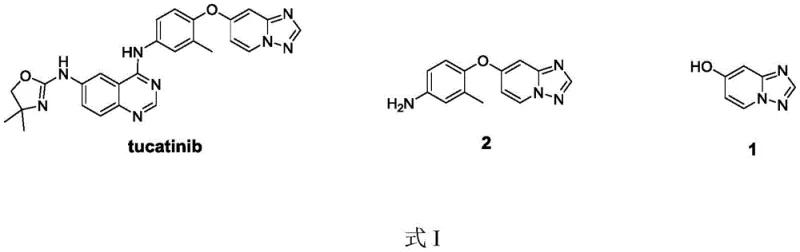
The structural complexity of Tucatinib necessitates robust and reliable sourcing of its precursors. As illustrated in the chemical architecture, the 7-hydroxy-[1,2,4]triazolo[1,5-a]pyridine moiety serves as the foundational scaffold for the subsequent etherification and coupling reactions required to assemble the final drug substance. Ensuring the consistent quality and economic viability of this intermediate is paramount for maintaining the continuity of Tucatinib production. The innovation presented in the referenced patent addresses historical bottlenecks in this supply chain by introducing a concise, high-yielding route that aligns with modern green chemistry principles and industrial scalability requirements.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this innovation, the synthesis of 7-hydroxy-[1,2,4]triazolo[1,5-a]pyridine was plagued by inefficient multi-step sequences that hindered cost-effective manufacturing. As depicted in the comparative reaction schemes, traditional Method I relied on the nucleophilic substitution of 2-nitro-4-chloropyridine with benzyl alcohol, followed by reduction and cyclization steps that ultimately required palladium-catalyzed hydrogenolysis to remove the benzyl protecting group. This route suffered from a dismal total yield of merely 20% over the final three steps and necessitated rigorous purification via column chromatography, a technique that is notoriously difficult to scale and economically prohibitive for bulk production. Furthermore, Method II utilized picolinic acid derivatives requiring four preliminary steps to generate the expensive 2-amino-4-methoxypyridine starting material, resulting in an overall yield of only 33% for the precursor alone.
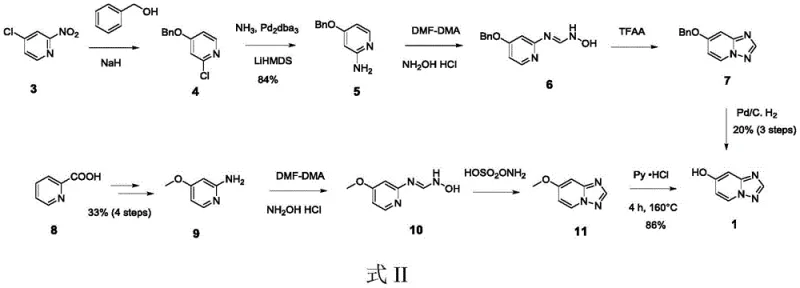
The reliance on protecting groups such as benzyl or methyl ethers in these conventional pathways introduces significant operational overhead. Each protection and deprotection cycle adds time, consumes additional reagents, and generates substantial chemical waste, thereby inflating the environmental footprint and production costs. The necessity for high-temperature melting steps or harsh demethylation conditions in Method II further compromises process safety and equipment longevity. These inherent inefficiencies create a fragile supply chain vulnerable to raw material price volatility and processing delays, making the search for a more direct and atom-economical synthesis not just a technical preference but a commercial imperative for pharmaceutical procurement teams seeking reliability.
The Novel Approach
In stark contrast to the convoluted legacy pathways, the novel methodology disclosed in CN114671867A streamlines the synthesis into a highly efficient two-step sequence that bypasses the need for any protecting groups. This innovative route initiates with the direct condensation of commercially abundant 2-amino-4-hydroxypyridine with N,N-dimethylformamide dimethyl acetal (DMF-DMA), followed by immediate conversion to the amidine intermediate using hydroxylamine hydrochloride. The subsequent intramolecular cyclization is elegantly achieved using trifluoroacetic anhydride (TFAA) under mild thermal conditions, directly furnishing the target 7-hydroxy-[1,2,4]triazolo[1,5-a]pyridine. This approach eliminates the tedious separation and purification steps associated with intermediate isolation, allowing the crude amidine to proceed directly to cyclization with minimal handling.
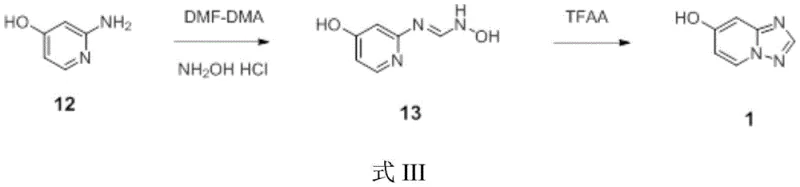
The operational simplicity of this new process is a game-changer for industrial scale-up. By removing the requirement for column chromatography and replacing it with straightforward crystallization and washing protocols, the method drastically reduces solvent consumption and processing time. The reaction conditions are notably mild, typically operating between 50°C and 95°C, which lowers energy demands and enhances safety profiles compared to the high-temperature operations of previous methods. Moreover, the use of inexpensive and readily available solvents like ethanol and tetrahydrofuran ensures that the process remains economically viable even at the multi-ton scale. This direct access to the heterocyclic core represents a significant technological leap, offering a robust platform for the sustainable manufacturing of oncology intermediates.
Mechanistic Insights into Direct Triazolopyridine Cyclization
The chemical elegance of this synthesis lies in the precise orchestration of amidine formation and oxidative cyclization. In the first stage, 2-amino-4-hydroxypyridine undergoes a formamidination reaction where the exocyclic amine attacks the electrophilic carbon of DMF-DMA, displacing dimethylamine to form an N-(4-hydroxypyridin-2-yl)-N',N'-dimethylformamidine species. This reactive intermediate is then treated with hydroxylamine hydrochloride, which facilitates a nucleophilic attack on the formamidine carbon, resulting in the displacement of dimethylamine and the formation of the N-hydroxy-N'-(4-hydroxypyridin-2-yl)-formamide intermediate. This step is critical as it installs the necessary nitrogen-oxygen functionality required for the subsequent ring closure, proceeding with exceptional efficiency to yield the intermediate with purity exceeding 99% without the need for intermediate purification.
The final cyclization step is driven by the activation of the formamide carbonyl by trifluoroacetic anhydride (TFAA). The electron-deficient carbonyl carbon becomes highly susceptible to intramolecular nucleophilic attack by the adjacent pyridine nitrogen or the hydroxylamine nitrogen, depending on the specific tautomeric state, ultimately leading to the formation of the fused [1,2,4]triazolo ring system. The use of TFAA serves a dual purpose: it acts as a dehydrating agent to drive the equilibrium towards the cyclized product and activates the substrate for ring closure under relatively mild conditions. This mechanism avoids the harsh acidic or basic conditions often required for similar heterocycle formations, thereby preserving the integrity of the sensitive hydroxyl group on the pyridine ring and minimizing the formation of degradation by-products or polymeric impurities.
![Chemical structure of the target intermediate 7-hydroxy-[1,2,4]triazolo[1,5-a]pyridine](/insights/img/tucatinib-intermediate-synthesis-supplier-pharma-20260305010728-03.png)
Impurity control in this process is inherently superior due to the high selectivity of the cyclization reaction. The absence of protecting groups eliminates the risk of incomplete deprotection or side reactions associated with protecting group manipulation, which are common sources of difficult-to-remove impurities in traditional routes. The workup procedure, involving neutralization with weak bases like sodium bicarbonate and simple recrystallization from alcohols, effectively removes residual acids, salts, and organic by-products. This results in a final product with purity levels consistently above 98%, as confirmed by HPLC analysis in the patent examples. Such high purity is essential for downstream coupling reactions in the synthesis of Tucatinib, where trace impurities could otherwise poison catalysts or complicate the final API purification, ensuring a smoother path to regulatory approval.
How to Synthesize 7-hydroxy-[1,2,4]triazolo[1,5-a]pyridine Efficiently
The practical execution of this synthesis is designed for seamless integration into existing pharmaceutical manufacturing facilities. The process begins with the suspension of 2-amino-4-hydroxypyridine in an alcoholic solvent such as ethanol or methanol, followed by the addition of DMF-DMA and heating to reflux to ensure complete conversion to the formamidine. Upon cooling, hydroxylamine hydrochloride is introduced to generate the key amidine intermediate, which can be isolated or used in situ. The subsequent cyclization involves dissolving this intermediate in a polar aprotic solvent like THF and treating it with TFAA at controlled temperatures ranging from 50°C to 75°C. The detailed standardized operating procedures for reaction times, stoichiometry, and workup protocols are critical for maximizing yield and reproducibility.
- React 2-amino-4-hydroxypyridine with DMF-DMA in alcohol solvent at reflux, followed by treatment with hydroxylamine hydrochloride to form the amidine intermediate.
- Perform intramolecular cyclization using trifluoroacetic anhydride (TFAA) in THF or acetonitrile under mild heating conditions.
- Isolate the final product through simple aqueous workup and recrystallization, achieving high purity without column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthetic route offers profound strategic benefits that extend beyond mere technical feasibility. The elimination of multiple synthetic steps and the removal of expensive protecting group chemistries translate directly into a leaner, more cost-efficient manufacturing process. By avoiding the use of precious metal catalysts like palladium, which are subject to significant market price fluctuations and require complex recovery systems, the new method stabilizes raw material costs. Furthermore, the simplification of the purification process from column chromatography to simple filtration and washing drastically reduces solvent usage and waste disposal costs, contributing to a lower overall cost of goods sold (COGS) and a smaller environmental footprint.
- Cost Reduction in Manufacturing: The streamlined two-step process significantly lowers production expenses by reducing reagent consumption and eliminating the need for costly chromatographic purification media. The avoidance of benzyl or methyl protecting groups removes entire sequences of protection and deprotection reactions, which traditionally account for a substantial portion of manufacturing time and material costs. Additionally, the high yields reported in the patent examples, reaching up to 87% for the intermediate and 81% for the final product, ensure maximum material throughput and minimize waste generation. This efficiency allows for competitive pricing strategies in the supply of high-purity pharmaceutical intermediates, providing a distinct market advantage.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals such as 2-amino-4-hydroxypyridine, DMF-DMA, and hydroxylamine hydrochloride ensures a robust and resilient supply chain less susceptible to disruptions. Unlike specialized starting materials that may have limited suppliers or long lead times, the raw materials for this process are widely available from multiple global vendors. The simplified process flow also reduces the risk of batch failures associated with complex multi-step syntheses, ensuring consistent on-time delivery to API manufacturers. This reliability is crucial for maintaining uninterrupted production schedules for critical oncology medications like Tucatinib, safeguarding patient access to essential therapies.
- Scalability and Environmental Compliance: The mild reaction conditions and simple workup procedures make this process exceptionally well-suited for commercial scale-up from pilot plant to multi-ton production. The absence of hazardous reagents and the reduction in solvent intensity align with increasingly stringent environmental regulations and corporate sustainability goals. The ability to produce high-purity material without generating heavy metal waste or excessive organic solvent waste simplifies regulatory compliance and reduces the burden on waste treatment facilities. This scalability ensures that the supply can easily expand to meet growing global demand for HER2 inhibitors without the need for massive capital investment in specialized equipment.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. Understanding these details is vital for R&D teams evaluating process transfer and procurement officers assessing supplier capabilities. The answers are derived directly from the experimental data and technical specifications provided in the patent literature, ensuring accuracy and relevance for decision-making processes.
Q: What are the primary advantages of the new synthesis route over prior art methods?
A: The novel route eliminates the need for benzyl or methyl protecting groups, reduces the step count significantly, and avoids expensive column chromatography purification, leading to higher overall yields and lower production costs.
Q: Is the process suitable for large-scale industrial manufacturing?
A: Yes, the method utilizes readily available raw materials like 2-amino-4-hydroxypyridine and employs mild reaction conditions with simple workup procedures, making it highly amenable to multi-kilogram and ton-scale production.
Q: What purity levels can be achieved with this methodology?
A: The process consistently delivers intermediates with purity exceeding 99% after the first step and final product purity greater than 98% after simple washing and drying, meeting stringent pharmaceutical standards.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 7-hydroxy-[1,2,4]triazolo[1,5-a]pyridine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient intermediate synthesis in the global fight against cancer. Our technical team has thoroughly analyzed the advancements presented in patent CN114671867A and is fully equipped to leverage this chemistry for your supply needs. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and compliant. Our state-of-the-art facilities are designed to handle complex heterocyclic chemistry with precision, adhering to stringent purity specifications and rigorous QC labs to guarantee that every batch meets the highest international standards for pharmaceutical intermediates.
We invite you to collaborate with us to optimize your supply chain for Tucatinib and related HER2 inhibitors. By partnering with our expert technical procurement team, you can gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact us today to request specific COA data and route feasibility assessments, allowing us to demonstrate how our advanced manufacturing capabilities can drive value and security into your pharmaceutical supply chain.