Advanced Four-Step Synthesis of Jaspamycin for Commercial Scale-Up
Introduction to the Streamlined Synthesis of Jaspamycin
The pharmaceutical industry is constantly seeking more efficient pathways for the production of bioactive nucleoside analogs, and the recent disclosure in patent CN112851719A presents a transformative approach to synthesizing Jaspamycin (CAS: 22242-96-2). This specific intellectual property addresses the critical bottlenecks of traditional manufacturing, namely the excessive number of reaction steps and the prohibitive costs associated with purification. By leveraging a concise four-step sequence that bypasses the need for column chromatography, this technology offers a compelling value proposition for reliable pharmaceutical intermediate suppliers aiming to optimize their production lines. The methodology not only simplifies the operational workflow but also enhances the overall safety profile by eliminating hazardous processes such as high-pressure hydrogenation or diazotization. For R&D directors and process chemists, this patent represents a significant leap forward in process intensification, providing a clear roadmap for generating high-purity material with improved economic efficiency.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of complex nucleoside derivatives like Jaspamycin has been plagued by inefficient multi-step sequences that rely heavily on labor-intensive purification techniques. Conventional routes often necessitate the use of expensive protecting group strategies that require multiple installation and removal cycles, each adding to the overall cost and reducing the cumulative yield. Furthermore, traditional methods frequently employ column chromatography for intermediate isolation, a technique that is notoriously difficult to scale due to solvent consumption, silica gel waste, and extended processing times. These factors collectively contribute to a high cost of goods sold (COGS) and create significant supply chain vulnerabilities, making it challenging for manufacturers to meet the demands of large-scale clinical or commercial programs. The reliance on dangerous reagents in older protocols also imposes strict regulatory and safety burdens on production facilities, further complicating the manufacturing landscape.
The Novel Approach
In stark contrast, the novel approach detailed in the patent utilizes a direct and atom-economical strategy that streamlines the construction of the target molecule through a highly optimized four-step cascade. The process initiates with a robust glycosylation reaction followed by sequential deprotection and functionalization steps that are carefully tuned to allow for simple isolation via pulping and crystallization. This elimination of chromatographic purification is a game-changer for cost reduction in pharmaceutical intermediate manufacturing, as it drastically reduces solvent usage and processing time. The route employs readily available starting materials, such as 4-chloro-5-iodo-7H-pyrrolo[2,3-d]pyrimidine, ensuring a stable supply chain and predictable pricing structures. By focusing on crystallization-driven purity, the method ensures that the final product meets stringent quality specifications without the need for complex downstream processing.
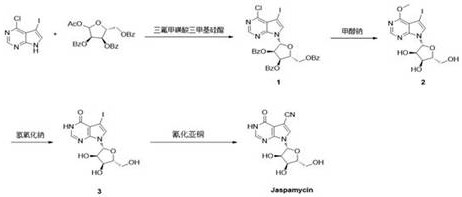
Mechanistic Insights into Silylated Glycosylation and Cyanation
The cornerstone of this synthetic route is the initial coupling reaction, which employs N,O-bis(trimethylsilyl)acetamide (BSA) and trimethylsilyl trifluoromethanesulfonate (TMSOTf) to activate the nucleobase for glycosylation. This silylation mechanism effectively increases the nucleophilicity of the pyrrolopyrimidine nitrogen, facilitating a stereoselective attack on the anomeric center of the protected ribofuranose donor. The reaction is conducted at elevated temperatures between 75°C and 85°C, typically optimized at 80°C, to ensure complete conversion while minimizing side reactions. This specific catalytic system is superior to traditional Lewis acid promoters because it generates fewer acidic byproducts that could degrade the sensitive sugar moiety, thereby preserving the integrity of the intermediate Compound 1. The high yield of 90% in this first step sets a strong foundation for the overall efficiency of the synthesis, demonstrating the power of modern silyl-mediated coupling chemistry.
Following the coupling, the process moves through a series of deprotection and functionalization steps culminating in a Rosenmund-von Braun type cyanation. The final transformation involves heating Compound 3 with cuprous cyanide in pyridine at temperatures ranging from 100°C to 115°C. This step is critical for introducing the nitrile functionality essential for the biological activity of Jaspamycin. The mechanism likely proceeds through a copper-mediated nucleophilic aromatic substitution where the iodine leaving group is displaced by the cyanide ion. Crucially, the patent highlights that the crude product from this step can be purified simply by pulping with aqueous ammonia and recrystallization, indicating that the reaction selectivity is high enough to prevent the formation of difficult-to-remove impurities. This mechanistic understanding allows process engineers to tightly control reaction parameters to maximize yield and minimize waste.
How to Synthesize Jaspamycin Efficiently
Implementing this synthesis requires precise control over reaction temperatures and stoichiometry to replicate the high yields reported in the patent data. The process begins with the silylation of the heterocycle in acetonitrile, followed by the addition of the sugar donor and catalyst, requiring a sustained heat period of approximately 18 hours. Subsequent steps involve careful pH control during the hydrolysis phase and strict temperature management during the cyanation to prevent decomposition. The detailed standardized synthesis steps below outline the specific conditions required to achieve the reported 36.5% total yield while maintaining operational simplicity.
- Couple 4-chloro-5-iodo-7H-pyrrolo[2,3-d]pyrimidine with protected ribofuranose using BSA and TMSOTf at 80°C to form Compound 1.
- Perform deacetylation on Compound 1 using sodium methoxide in methanol at 25°C to obtain Compound 2 directly without purification.
- Hydrolyze Compound 2 in aqueous sodium hydroxide under reflux to generate Compound 3, followed by acid neutralization and filtration.
- React Compound 3 with cuprous cyanide in pyridine at 115°C, followed by pulping and recrystallization to yield pure Jaspamycin.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis route offers profound strategic advantages that extend far beyond simple chemical transformation. The most significant benefit lies in the drastic simplification of the purification workflow, which directly translates to substantial cost savings in manufacturing operations. By removing the requirement for column chromatography, facilities can reduce their solvent inventory needs, lower waste disposal costs, and significantly shorten the batch cycle time. This efficiency gain allows for a higher throughput of material within existing infrastructure, effectively increasing capacity without the need for capital-intensive equipment upgrades. Furthermore, the use of commodity chemicals and the avoidance of specialized, high-cost reagents ensure that the raw material supply remains stable and resilient against market fluctuations.
- Cost Reduction in Manufacturing: The economic impact of this process is driven primarily by the elimination of chromatographic purification, which is traditionally one of the most expensive unit operations in fine chemical synthesis. Without the need for large quantities of silica gel and elution solvents, the variable costs per kilogram of product are significantly lowered. Additionally, the high yield of the initial coupling step (90%) ensures that expensive starting materials are utilized with maximum efficiency, reducing the cost of wasted inputs. The ability to isolate intermediates via simple filtration and pulping further reduces labor costs and energy consumption associated with rotary evaporation and drying large solvent volumes.
- Enhanced Supply Chain Reliability: The reliance on widely available starting materials such as 4-chloro-5-iodo-7H-pyrrolo[2,3-d]pyrimidine and protected ribose derivatives ensures a robust supply chain that is less susceptible to single-source bottlenecks. Unlike routes that depend on exotic catalysts or custom-synthesized precursors, this method utilizes reagents that are commercially accessible from multiple global vendors. This diversification of the supply base mitigates the risk of production delays caused by raw material shortages. Moreover, the simplified process flow reduces the complexity of logistics, allowing for faster turnaround times from order placement to delivery of the final active pharmaceutical ingredient intermediate.
- Scalability and Environmental Compliance: From an environmental and regulatory standpoint, this route is exceptionally well-suited for commercial scale-up due to its reduced solvent intensity and avoidance of hazardous unit operations. The absence of high-pressure hydrogenation or diazotization reactions lowers the safety risk profile of the plant, simplifying regulatory compliance and insurance requirements. The process generates less chemical waste, particularly solid waste from silica gel, aligning with modern green chemistry principles and sustainability goals. This environmental friendliness not only reduces disposal costs but also enhances the corporate social responsibility profile of the manufacturing entity, making it a preferred partner for environmentally conscious pharmaceutical clients.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Jaspamycin synthesis technology. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on yield expectations, purification methods, and scalability potential. Understanding these details is crucial for technical teams evaluating the feasibility of adopting this route for their own production pipelines.
Q: What are the primary advantages of this new Jaspamycin synthesis method over conventional routes?
A: The patented method significantly reduces the number of reaction steps to just four and eliminates the need for expensive and time-consuming column chromatography purification. By utilizing cheap starting materials and avoiding dangerous reactions like hydrogenation, it offers a safer and more cost-effective pathway suitable for mass production.
Q: How does the process ensure high purity without chromatographic separation?
A: High purity is achieved through a strategic combination of selective precipitation, pulping, and recrystallization techniques at critical intermediate stages. Specifically, Compound 1 and the final product are isolated via pulping and crystallization, which effectively removes impurities while maintaining a robust total yield of approximately 36.5%.
Q: Is this synthesis route scalable for industrial manufacturing of nucleoside intermediates?
A: Yes, the route is explicitly designed for scalability by avoiding hazardous unit operations and utilizing standard heating and stirring conditions. The use of common solvents like acetonitrile, methanol, and pyridine, along with the elimination of complex purification steps, makes it highly adaptable for commercial scale-up from kilogram to tonnage levels.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Jaspamycin Supplier
At NINGBO INNO PHARMCHEM, we recognize the immense potential of this optimized synthetic route to revolutionize the availability of high-quality Jaspamycin for the global market. As a leading CDMO expert, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from laboratory bench to full-scale manufacturing. Our facility is equipped with rigorous QC labs and adheres to stringent purity specifications, guaranteeing that every batch of nucleoside intermediate meets the highest international standards. We are committed to leveraging our technical expertise to refine this process further, optimizing yields and minimizing environmental impact to deliver superior value to our partners.
We invite you to collaborate with us to unlock the full commercial potential of this technology. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating exactly how this route can improve your bottom line. Please contact us today to request specific COA data and route feasibility assessments, and let us demonstrate why we are the preferred partner for your complex nucleoside synthesis needs.