Advanced Manufacturing of Kinase Inhibitor Intermediates via Optimized Suzuki-Miyaura Coupling
Introduction to Patent CN103153956A
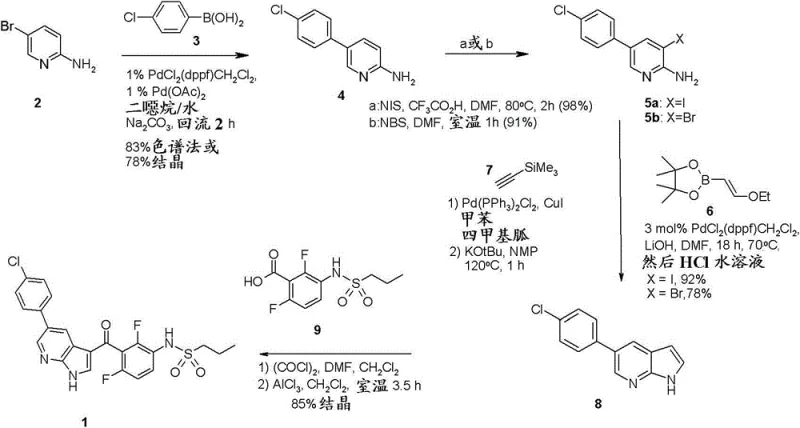
The pharmaceutical industry continuously seeks robust and scalable synthetic routes for complex kinase inhibitors, particularly those targeting the BRAF pathway. Patent CN103153956A discloses a groundbreaking methodology for the preparation of propane-1-sulfonic acid {3-[5-(4-chloro-phenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl]-2,4-difluoro-phenyl}-amide, a critical intermediate in the synthesis of oncology therapeutics like Vemurafenib. This intellectual property represents a significant leap forward in process chemistry, addressing long-standing challenges related to yield optimization, step economy, and operational safety. By leveraging advanced transition metal catalysis, specifically dual Suzuki-Miyaura coupling strategies, the inventors have established a pathway that not only enhances the purity profile of the final product but also streamlines the manufacturing workflow. For R&D directors and procurement specialists, understanding the nuances of this patent is essential for securing a reliable supply chain for high-value pharmaceutical intermediates.
The core innovation lies in the construction of the 5-substituted-7-azaindole scaffold, a privileged structure in medicinal chemistry. Traditional methods often suffer from low regioselectivity or require hazardous reagents that complicate large-scale production. In contrast, the approach detailed in CN103153956A utilizes a modular sequence starting from readily available aminopyridines. The process integrates a initial biaryl coupling, followed by selective halogenation, and culminates in a sophisticated cyclization step that builds the pyrrole ring onto the pyridine core. This strategic disconnection allows for greater flexibility in sourcing starting materials and provides multiple off-ramps for quality control, ensuring that the final active pharmaceutical ingredient (API) meets stringent regulatory standards for impurity levels.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historical synthetic routes for 7-azaindole derivatives have frequently relied on condensation reactions that demand harsh thermal conditions or strong bases, leading to significant degradation of sensitive functional groups. Furthermore, the preparation of necessary vinyl boronate reagents, which are crucial for constructing the five-membered ring, has traditionally involved the use of ethoxyacetylene. This reagent is highly flammable and poses substantial explosion risks during storage and handling, creating a major bottleneck for chemical manufacturers aiming for safe commercial scale-up. Additionally, older methodologies often involve multiple purification steps between intermediates, which drastically reduces the overall mass balance and increases the cost of goods sold (COGS) due to solvent consumption and extended processing times. These inefficiencies make conventional routes less attractive for modern, lean manufacturing environments focused on sustainability and cost reduction.
The Novel Approach
The methodology presented in this patent circumvents these issues by introducing a safer and more efficient catalytic cycle for generating the key vinyl boronate species. Instead of relying on dangerous acetylene derivatives, the invention employs a dehydroborylation of ethyl vinyl ether using palladium, rhodium, or ruthenium catalysts. This transformation proceeds under mild conditions with high selectivity, effectively eliminating the safety hazards associated with previous methods. Moreover, the synthetic sequence for the final amide utilizes a Friedel-Crafts acylation that is optimized for room temperature execution, reducing energy consumption. The ability to choose between a Suzuki-Miyaura pathway or a Sonogashira pathway for the ring-closing step provides process chemists with valuable redundancy; if one catalyst system faces supply constraints, the alternative route ensures continuity of supply. This flexibility is a critical asset for supply chain heads managing risk in the global pharmaceutical market.
Mechanistic Insights into Pd-Catalyzed Cross-Coupling and Cyclization
The heart of this synthetic strategy is the precise orchestration of palladium-catalyzed cross-coupling reactions. The initial step involves the oxidative addition of a Pd(0) species into the carbon-bromine bond of the aminopyridine substrate, forming a reactive organopalladium intermediate. This is followed by transmetallation with the aryl boronic acid, facilitated by a base such as sodium carbonate or lithium hydroxide, which activates the boron species for nucleophilic attack. The subsequent reductive elimination releases the biaryl product and regenerates the active catalyst, completing the cycle. What distinguishes this process is the tolerance of the catalyst system to the free amino group on the pyridine ring, which typically acts as a poison to palladium catalysts. The patent specifies ligand systems, such as dppf, that stabilize the metal center and prevent catalyst deactivation, ensuring high turnover numbers and consistent reaction kinetics throughout the batch.
Following the biaryl formation, the introduction of a halogen atom (iodine or bromine) at the 3-position of the pyridine ring is achieved via electrophilic aromatic substitution using N-halosuccinimides. This activation is crucial for the subsequent ring-closing reaction. In the preferred Suzuki-Miyaura variant, the halogenated intermediate reacts with a vinyl boronate ester. The resulting vinyl-aminopyridine undergoes an intramolecular cyclization upon treatment with acid. Mechanistically, the acid protonates the vinyl ether or activates the alkene, making it susceptible to nucleophilic attack by the adjacent amino group. This cascade forms the five-membered pyrrole ring, aromatizing to yield the stable 7-azaindole core. The alternative Sonogashira route utilizes a copper co-catalyst to couple a silylated acetylene, followed by base-mediated desilylation and cyclization, offering a mechanistically distinct but equally effective pathway to the same structural motif.
How to Synthesize 5-Substituted-7-Azaindole Efficiently
The practical execution of this synthesis requires careful attention to reaction parameters to maximize the benefits described in the patent. The process begins with the coupling of 2-amino-5-bromopyridine and 4-chlorophenylboronic acid in a biphasic solvent system, typically dioxane and water, maintained at elevated temperatures to drive conversion. Following isolation of the biaryl amine, the halogenation step is performed in polar aprotic solvents like DMF, where stoichiometry must be tightly controlled to prevent di-halogenation. The critical ring-closing step involves the use of the novel vinyl boronate reagent, which can be prepared in situ or sourced as a stable intermediate. The final cyclization is triggered by the addition of aqueous hydrochloric acid, which simultaneously hydrolyzes the vinyl ether and promotes ring closure. Detailed standardized synthetic steps for laboratory and pilot plant execution are provided below to ensure reproducibility and safety compliance.
- Perform a initial Suzuki-Miyaura coupling between 2-amino-5-bromopyridine and 4-chlorophenylboronic acid using a palladium catalyst system.
- Execute a halogenation step using N-iodosuccinimide (NIS) or N-bromosuccinimide (NBS) to activate the pyridine ring for further functionalization.
- Conduct a second coupling reaction with a vinyl boronate ester followed by acid-mediated cyclization to form the 7-azaindole core structure.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of the technology described in CN103153956A offers tangible economic and logistical benefits beyond simple chemical yield. The most significant advantage is the radical improvement in process safety regarding the synthesis of vinyl boronate reagents. By replacing highly flammable ethoxyacetylene with stable ethyl vinyl ether and a palladium catalyst, the inventory risk and insurance costs associated with raw material storage are drastically reduced. This shift eliminates the need for specialized explosion-proof infrastructure, allowing for production in standard chemical facilities and thereby lowering capital expenditure requirements for contract manufacturing organizations (CMOs). Furthermore, the high yields reported in the patent examples, such as 98% for the iodination step and 92% for the cyclization, translate directly into reduced waste generation and lower disposal costs, aligning with green chemistry principles and environmental regulations.
From a supply continuity perspective, the modularity of this route is a game-changer. The ability to source either iodine or bromine intermediates allows procurement teams to hedge against volatility in halogen markets. If iodine prices spike due to geopolitical factors, the process can seamlessly switch to the bromine-based pathway without requiring a complete re-validation of the manufacturing line. Additionally, the use of commodity chemicals like 2-amino-5-bromopyridine and 4-chlorophenylboronic acid ensures that the supply chain is not dependent on exotic or single-source custom synthons. This diversification of the supplier base enhances resilience against disruptions. The simplified work-up procedures, often involving simple precipitation or crystallization rather than complex chromatography, further reduce the lead time for batch release, enabling faster response to market demand fluctuations for the final oncology drug.
- Cost Reduction in Manufacturing: The elimination of hazardous reagents and the reduction in purification steps significantly lower the operational expenditure per kilogram of produced intermediate. By avoiding expensive chromatographic separations and utilizing high-yielding catalytic steps, the overall material cost is optimized. The process minimizes solvent usage through efficient precipitation techniques, reducing both purchase and recycling costs. Furthermore, the higher throughput achieved by shorter reaction times and milder conditions allows existing reactor capacity to produce more volume, effectively spreading fixed costs over a larger output and driving down the unit price for the pharmaceutical intermediate.
- Enhanced Supply Chain Reliability: The reliance on stable, non-hazardous starting materials like ethyl vinyl ether instead of explosive acetylenes removes a critical vulnerability from the supply chain. This stability ensures that raw material deliveries are less likely to be delayed by safety inspections or transport restrictions. The robustness of the palladium catalyst systems described ensures consistent batch-to-batch quality, reducing the risk of production failures that could halt downstream API synthesis. This reliability is paramount for maintaining the continuous supply of life-saving cancer medications, where interruptions can have severe clinical consequences.
- Scalability and Environmental Compliance: The process is designed with scale-up in mind, utilizing reagents and conditions that are well-understood in multi-ton reactors. The avoidance of cryogenic temperatures and the use of ambient pressure reactions simplify engineering requirements. Environmentally, the reduction in waste streams due to higher atom economy and fewer purification steps lowers the facility's environmental footprint. This compliance with stricter environmental standards future-proofs the manufacturing site against evolving regulations, ensuring long-term operational viability without the need for costly retrofits or waste treatment upgrades.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis route. These insights are derived directly from the experimental data and claims within CN103153956A, providing a factual basis for decision-making. Understanding these details helps stakeholders evaluate the feasibility of adopting this technology for their specific production needs. The answers cover aspects ranging from catalyst selection to impurity management, ensuring a comprehensive overview of the process capabilities.
Q: What are the safety advantages of the new vinyl boronate synthesis method?
A: The patent describes a dehydroborylation method using palladium catalysts on ethyl vinyl ether, which avoids the use of highly flammable and hazardous ethoxyacetylene required in traditional methods, significantly improving industrial safety profiles.
Q: How does this process improve overall yield compared to conventional routes?
A: By minimizing isolation steps and utilizing high-yielding coupling reactions (up to 98% for halogenation and 92% for cyclization), the process reduces material loss and improves the overall throughput of the final API intermediate.
Q: Is this synthesis route scalable for commercial production?
A: Yes, the use of robust palladium catalysts, standard solvents like DMF and toluene, and the avoidance of cryogenic conditions make this route highly suitable for scale-up from kilogram to multi-ton commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Propane-1-sulfonic acid amide Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and safe synthetic routes in the production of high-value pharmaceutical intermediates. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the innovative chemistry described in patent CN103153956A can be translated into reality for your supply chain. Our state-of-the-art facilities are equipped to handle the specific palladium-catalyzed reactions and strict purity specifications required for kinase inhibitor intermediates. With our rigorous QC labs and commitment to process excellence, we guarantee that every batch meets the highest international standards for purity and safety, mitigating risk for your downstream API manufacturing.
We invite you to collaborate with us to leverage this advanced technology for your oncology drug programs. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this route can optimize your COGS. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. By partnering with NINGBO INNO PHARMCHEM, you secure not just a supplier, but a strategic ally dedicated to advancing your pharmaceutical development timelines through superior chemical manufacturing solutions.