Scalable Synthesis of Alpha-Aza Spirocyclic Drug Templates for Commercial API Production
Scalable Synthesis of Alpha-Aza Spirocyclic Drug Templates for Commercial API Production
The development of efficient synthetic routes for complex heterocyclic scaffolds remains a critical bottleneck in modern pharmaceutical research, particularly for structures serving as Type II beta-turn mimics. Patent CN101081851A discloses a groundbreaking preparation method for alpha-aza spirocyclic drug templates that addresses the longstanding challenges of low yield and excessive step count associated with traditional syntheses. This technology enables the rapid construction of diverse spirocyclic libraries, such as [5.5], [5.6], [6.5], and [6.6] systems, which are essential for optimizing the pharmacokinetic profiles of peptide-based therapeutics and small molecule drugs. By streamlining the synthesis into just four robust chemical transformations, this innovation offers a reliable pharmaceutical intermediate supplier pathway for generating high-purity scaffolds needed for combinatorial chemistry and lead optimization campaigns.
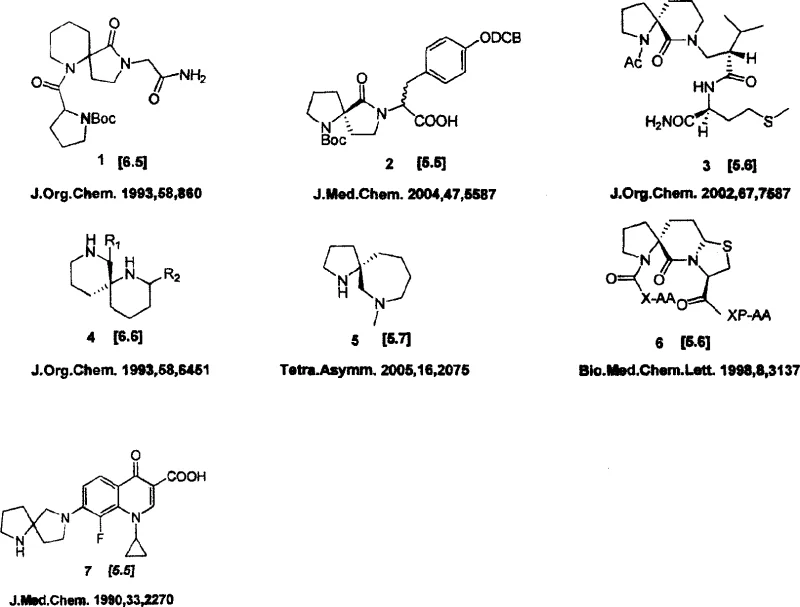
The strategic value of these alpha-aza spirocyclic cores lies in their ability to constrain conformational flexibility, thereby enhancing binding affinity and metabolic stability in drug candidates. As illustrated in the structural diversity shown in the patent documentation, these templates can be functionalized at multiple positions to create extensive compound libraries. The ability to access these structures efficiently allows research teams to accelerate the discovery of novel antibiotics, such as quinolone derivatives, and other bioactive agents where spirocyclic substitution has been proven to markedly improve physiological activity. This patent represents a significant leap forward in process chemistry, transforming a previously arduous multi-step sequence into a streamlined, industrially viable protocol.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this invention, the synthesis of alpha-aza spirocyclic templates was plagued by inefficient methodologies that hindered large-scale production. As detailed in the background art, existing methods such as those reported in J.Med.Chem.1990 and J.Org.Chem.1993 typically required between five to eight distinct reaction steps to construct the spirocyclic core. These legacy routes often relied on harsh reagents, expensive protecting group manipulations, and low-yielding ring-closing reactions, resulting in dismal overall yields ranging from merely 4% to 21%. For instance, Method 1 utilized a sequence involving lithium diisopropylamide (LDA) alkylation followed by reduction and aminolysis, yet suffered from a cumulative yield of only 7%. Similarly, Method 3 employed a complex sequence of allylation, ozonolysis, and Mitsunobu-type cyclization, achieving a mere 4% overall yield. Such inefficiencies not only drive up the cost of goods but also generate substantial chemical waste, making these routes economically unfeasible for commercial API manufacturing.
The Novel Approach
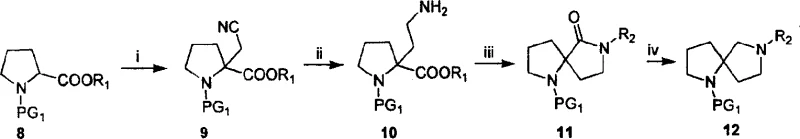
In stark contrast to the convoluted pathways of the past, the novel approach described in CN101081851A simplifies the synthesis to a concise four-step sequence: amidation, oxidation, alkylation, and hydrogenative cyclization. This methodology starts from readily available proline or pipecolic acid derivatives carrying a protected amino group, eliminating the need for complex starting materials. The key innovation lies in the final step, where a nitrile-containing intermediate undergoes catalytic hydrogenation in the presence of ammonia to simultaneously reduce the nitrile and close the ring, forming the spirocyclic amine in high yield. This strategy bypasses the need for separate reduction and cyclization steps, drastically shortening the synthetic timeline. The general formula of the resulting compounds demonstrates the versatility of this approach, allowing for the modulation of ring sizes (m=1 or 2) and side chains (n=1, 2, or 3) to tailor the scaffold for specific biological targets.
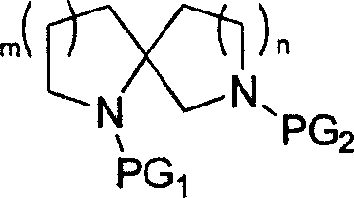
Mechanistic Insights into the Four-Step Spirocyclization Strategy
The success of this synthetic route hinges on the precise control of reactivity at the alpha-position of the pyrrolidine or piperidine ring. The process initiates with the conversion of the carboxylic acid to a primary amide using active esters like vinyl chloroformate, followed by dehydration to a nitrile using trifluoroacetic anhydride (TFAA). The critical mechanistic step occurs during the alkylation phase, where the alpha-proton of the nitrile is abstracted by a strong base, specifically LDA, at cryogenic temperatures (-78°C). This generates a stabilized carbanion that attacks a di-haloalkane, such as 1-bromo-2-chloroethane, introducing the necessary carbon chain for ring closure. The choice of di-haloalkane dictates the size of the second ring in the spiro system, providing a modular handle for structural diversification. The subsequent hydrogenation step is equally sophisticated; under hydrogen pressure (30-45 psi) and elevated temperature (40-65°C) with a Raney Nickel catalyst, the nitrile is reduced to a primary amine which immediately undergoes intramolecular nucleophilic substitution with the terminal halide to form the stable spirocyclic structure.
![Embodiment 1 reaction scheme showing the 4-step synthesis of 1,7-diazaspiro[4.4]nonane](/insights/img/alpha-aza-spirocyclic-template-synthesis-pharma-supplier-20260307111058-09.webp)
Impurity control is inherently built into this mechanism through the use of robust protecting groups and selective reagents. The use of Boc (tert-butyloxycarbonyl) protection ensures that the nitrogen atom remains inert during the harsh alkylation conditions, preventing polymerization or side reactions. Furthermore, the oxidative dehydration step using TFAA is highly selective for the primary amide, minimizing the formation of over-oxidized byproducts. The final hydrogenation step, conducted in alcoholic solvents with added ammonia, serves a dual purpose: it reduces the nitrile while the ammonia suppresses secondary amine formation, ensuring the formation of the desired primary amine intermediate that cyclizes cleanly. This mechanistic elegance results in intermediates that are easier to purify, often requiring only simple extraction or crystallization rather than tedious chromatography, which is a major advantage for process chemistry.
How to Synthesize Alpha-Aza Spirocyclic Templates Efficiently
The synthesis of these valuable scaffolds is designed for operational simplicity, utilizing standard laboratory equipment and commercially available reagents. The procedure begins with the activation of N-Boc-proline followed by ammonolysis, proceeds through dehydration to the nitrile, and then employs low-temperature lithiation for chain extension. The final cyclization is achieved via catalytic hydrogenation, a unit operation familiar to any process chemist. For a detailed breakdown of the specific reaction conditions, stoichiometry, and workup procedures required to replicate this high-yielding transformation, please refer to the standardized synthesis guide below.
- Amidation of protected proline or pipecolic acid derivatives using active esters and ammonia to form 2-carbamoyl intermediates.
- Oxidative dehydration using trifluoroacetic anhydride (TFAA) to convert the amide into a nitrile group.
- Deprotonation with LDA followed by alkylation with di-haloalkanes to introduce the side chain required for ring closure.
- Catalytic hydrogenation using Raney Nickel or Pd/C under ammonia atmosphere to effect simultaneous reduction and intramolecular cyclization.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route translates directly into enhanced operational efficiency and risk mitigation. By reducing the number of unit operations from nearly eight down to just four, the process significantly lowers the consumption of solvents, reagents, and energy, leading to substantial cost savings in raw material procurement. The elimination of exotic or hazardous reagents, such as osmium tetroxide or azodicarboxylates found in prior art methods, simplifies safety compliance and reduces the burden on environmental health and safety (EHS) departments. This streamlined approach ensures a more reliable supply of high-purity pharmaceutical intermediates, as fewer steps mean fewer opportunities for batch failure or yield erosion.
- Cost Reduction in Manufacturing: The drastic reduction in step count inherently lowers the cost of goods sold (COGS) by minimizing labor hours and equipment occupancy time. Unlike previous methods that suffered from cumulative yield losses, this route maintains high efficiency at each stage, with individual step yields often exceeding 80-90%. The use of cost-effective catalysts like Raney Nickel instead of precious metals like palladium or platinum for the final cyclization further drives down catalyst costs. Additionally, the simplified purification requirements reduce the volume of silica gel and solvents needed for chromatography, resulting in significant waste disposal savings.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals such as proline, pipecolic acid, and simple di-haloalkanes ensures that the supply chain is robust and less susceptible to geopolitical disruptions or vendor shortages. The mild reaction conditions, particularly the moderate hydrogenation pressures and temperatures, allow the process to be easily transferred between different manufacturing sites without requiring specialized high-pressure reactors. This flexibility enhances supply continuity, enabling manufacturers to scale production rapidly to meet fluctuating market demands for API intermediates without lengthy process re-validation.
- Scalability and Environmental Compliance: The process is designed with green chemistry principles in mind, avoiding the generation of toxic heavy metal waste associated with older cyclization methods. The aqueous workups and use of recyclable solvents like ethanol and methanol align with modern sustainability goals, facilitating easier regulatory approval for commercial manufacturing. The high atom economy of the hydrogenative cyclization step ensures that the majority of the starting material ends up in the final product, minimizing the environmental footprint. This makes the technology ideal for the commercial scale-up of complex pharmaceutical intermediates, supporting the transition from gram-scale discovery to multi-ton production.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this spirocyclic synthesis technology. These answers are derived directly from the experimental data and claims within the patent specification, providing clarity on the practical aspects of adopting this route for your specific project needs.
Q: What is the overall yield advantage of this new spirocyclization method compared to prior art?
A: While conventional methods reported in literature typically achieve overall yields between 4% and 21% over 5 to 8 steps, this patented process achieves an overall yield of 27% to 35% in only 4 steps, significantly reducing material loss.
Q: Can this synthesis be scaled for industrial production of API intermediates?
A: Yes, the process utilizes mild reaction conditions (e.g., hydrogenation at 30-45 psi and 40-65°C) and avoids expensive transition metal catalysts in the final step by using Raney Nickel, making it highly suitable for commercial scale-up.
Q: What types of spirocyclic scaffolds can be generated using this platform?
A: The method is versatile and can produce various ring sizes including [5.5], [5.6], [6.5], and [6.6] systems by varying the starting amino acid (proline vs. pipecolic acid) and the length of the di-haloalkane alkylating agent.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Alpha-Aza Spirocyclic Template Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality scaffolds play in accelerating drug discovery timelines. Our team of expert process chemists has extensively evaluated the technology disclosed in CN101081851A and possesses the capability to implement this 4-step synthesis at scale. We offer extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from benchtop to plant. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of alpha-aza spirocyclic template meets the exacting standards required for clinical and commercial applications.
We invite you to collaborate with us to leverage this advanced synthetic route for your next drug development program. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can optimize your supply chain and reduce your overall development costs.