Advanced Synthesis of Imatinib Key Intermediates via Efficient Acid-Catalyzed Pyrimidine Coupling
Advanced Synthesis of Imatinib Key Intermediates via Efficient Acid-Catalyzed Pyrimidine Coupling
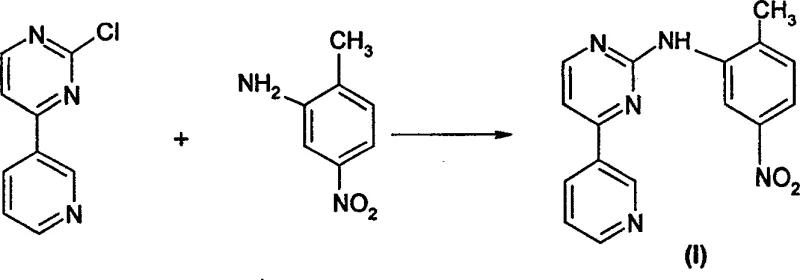
The pharmaceutical industry continuously seeks robust, scalable, and cost-effective pathways for the production of life-saving oncology medications, particularly tyrosine kinase inhibitors like Imatinib. Patent CN100537563C introduces a transformative organic synthesis methodology specifically designed for the preparation of N-(2-methyl-5-nitro)phenyl-4-(3-pyridyl)pyrimidin-2-amine, a critical precursor in the manufacture of Imatinib mesylate. This innovation addresses long-standing challenges in the supply chain of anti-leukemia therapeutics by replacing convoluted multi-step cyclization processes with a direct, high-yielding condensation reaction. For R&D directors and procurement specialists, this patent represents a pivotal shift towards more sustainable and economically viable manufacturing protocols, ensuring a stable supply of high-purity pharmaceutical intermediates essential for treating chronic myelogenous leukemia and gastrointestinal stromal tumors.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Imatinib and its structural analogs has relied on legacy processes described in earlier literature such as US5521184 and WO20040108699, which involve the reaction of 5-nitro-o-toluidine with 3-acetylpyridine. These traditional routes necessitate the initial formation of guanidine nitrate and beta-enone compounds, followed by a complex condensation and ring-closing sequence to generate the pyrimidine core. Such methodologies are plagued by significant operational drawbacks, including the requirement for hard-to-source starting materials in certain markets, the use of highly toxic reagents, and excessively long reaction sequences that degrade overall throughput. Furthermore, the harsh reaction conditions often associated with these older cyclization techniques lead to lower product yields and generate substantial chemical waste, creating bottlenecks for reliable pharmaceutical intermediate suppliers aiming to meet global demand efficiently.
The Novel Approach
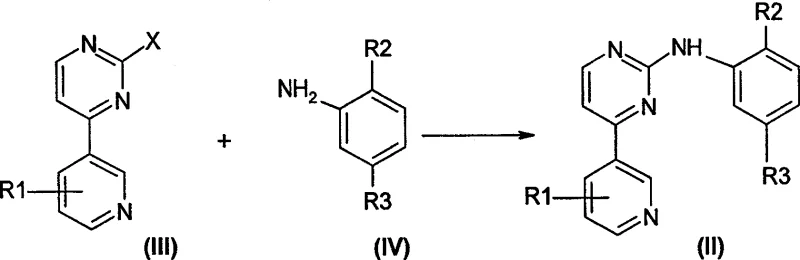
In stark contrast, the methodology disclosed in CN100537563C streamlines the production of N-phenyl-2-pyrimidinamine derivatives through a scientifically rationalized direct coupling strategy. This novel approach utilizes 4-aromatic heterocyclic group-2-halogenated pyrimidines (Structure III) and substituted anilines (Structure IV) as primary building blocks, reacting them in the presence of an acidic catalyst to form the target molecule in a single, efficient step. By bypassing the need for constructing the pyrimidine ring from scratch via guanidine intermediates, this process drastically reduces the number of unit operations and minimizes exposure to hazardous reagents. The result is a synthesis pathway that is not only shorter and safer but also delivers superior product yields, making it an ideal candidate for cost reduction in API manufacturing and large-scale industrial application.
Mechanistic Insights into Acid-Catalyzed Nucleophilic Substitution
The core chemical transformation driving this innovation is an acid-catalyzed nucleophilic aromatic substitution where the amino group of the substituted aniline attacks the electron-deficient carbon at the 2-position of the halopyrimidine ring. The presence of the electron-withdrawing nitrogen atoms in the pyrimidine ring activates the C-2 position towards nucleophilic attack, a process that is further accelerated by the protonation of the ring nitrogen or the leaving halogen group by the acidic catalyst. Suitable catalysts identified in the patent include strong inorganic acids like hydrochloric and sulfuric acid, as well as organic sulfonic acids such as methanesulfonic acid and p-toluenesulfonic acid, which facilitate the departure of the halide leaving group (preferably chlorine, bromine, or iodine). This mechanism allows the reaction to proceed smoothly in various anhydrous organic solvents like dioxane, THF, or acetonitrile, typically within a moderate temperature range of 50°C to 100°C, ensuring high selectivity and minimizing side reactions.
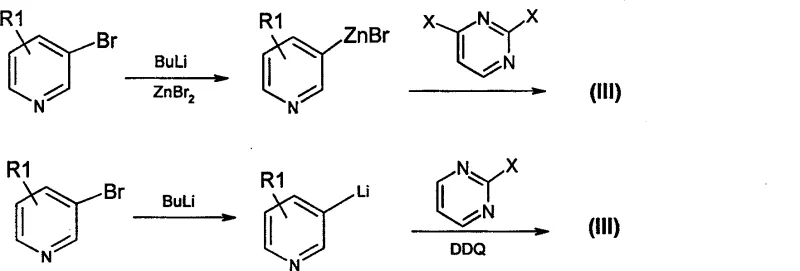
Furthermore, the synthesis of the crucial halopyrimidine reactant itself is optimized through advanced organometallic techniques, ensuring the availability of high-quality starting materials. As illustrated in the patent's Reaction Scheme B, 4-heteroaryl-2-halopyrimidines can be efficiently prepared via the lithiation or zincation of halopyridines followed by cross-coupling with dihalopyrimidines using palladium catalysis. This upstream process control is vital for maintaining the purity profile of the final intermediate, as impurities in the halopyrimidine precursor can propagate through the synthesis. By securing a robust supply of these specialized heterocyclic building blocks, manufacturers can ensure the consistent quality required for high-purity pharmaceutical intermediates, thereby reducing the burden on downstream purification steps like column chromatography or recrystallization.
How to Synthesize N-(2-methyl-5-nitro)phenyl-4-(3-pyridyl)pyrimidin-2-amine Efficiently
The practical implementation of this synthesis involves dissolving the 4-heteroaryl-2-halopyrimidine and the specific substituted aniline, such as p-nitro-o-toluidine for the Imatinib intermediate, in a suitable solvent like anhydrous dioxane. An acidic catalyst, such as methanesulfonic acid, is added to the mixture, which is then heated under reflux conditions for a defined period, typically around 6 hours, to drive the condensation to completion. Upon cooling, the reaction mixture is worked up by recovering the solvent and treating the residue with water and a base like sodium bicarbonate to precipitate the crude product, which is then collected and dried. While the general procedure is straightforward, precise control over stoichiometry, temperature, and workup pH is critical for maximizing yield and purity; therefore, the detailed standardized synthesis steps see the guide below.
- Preparation of 4-heteroaryl-2-halopyrimidine reactants via organolithium or organozinc cross-coupling with dihalopyrimidines.
- Condensation of the halopyrimidine with substituted aniline in an anhydrous organic solvent using an acidic catalyst.
- Reaction heating at 50-100°C followed by workup involving solvent recovery, basification, and crystallization to isolate the high-purity product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis route offers profound strategic benefits that extend beyond simple chemical efficiency. By transitioning from a multi-step cyclization process to a direct coupling reaction, manufacturers can significantly reduce the complexity of their production schedules, leading to shorter lead times and improved responsiveness to market fluctuations. The reliance on commercially available substituted anilines and the elimination of exotic, hard-to-source reagents mitigate supply chain risks, ensuring a more continuous and reliable flow of materials. This stability is crucial for maintaining the production of life-saving oncology drugs, where interruptions can have severe consequences for patients and healthcare providers globally.
- Cost Reduction in Manufacturing: The streamlined nature of this process inherently lowers manufacturing costs by reducing the number of reaction vessels, solvents, and energy inputs required per kilogram of product. Eliminating the need for synthesizing guanidine nitrate and beta-enone precursors removes entire stages of production, which translates to substantial savings in labor, utilities, and waste disposal. Additionally, the use of common acidic catalysts and standard organic solvents avoids the premium pricing associated with specialized reagents, further enhancing the economic viability of the process for large-scale commercial operations.
- Enhanced Supply Chain Reliability: The utilization of widely available starting materials, such as substituted anilines and dihalopyrimidines, ensures that the supply chain is not vulnerable to the bottlenecks often caused by niche chemical suppliers. This accessibility allows for the diversification of raw material sources, reducing dependency on single vendors and enhancing the overall resilience of the supply network. Consequently, partners can expect more consistent delivery schedules and a reduced risk of production stoppages due to material shortages, fostering a more stable partnership between suppliers and pharmaceutical companies.
- Scalability and Environmental Compliance: The reaction conditions, operating typically between 50°C and 100°C, are mild enough to be easily scaled from laboratory benchtop to multi-ton industrial reactors without requiring exotic high-pressure or cryogenic equipment. This ease of scale-up facilitates the commercial scale-up of complex pharmaceutical intermediates, allowing manufacturers to rapidly increase capacity to meet surging demand. Moreover, the simplified workflow generates less chemical waste and avoids the use of highly toxic intermediates, aligning with increasingly stringent environmental regulations and supporting sustainable manufacturing practices.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology, derived directly from the patent specifications and experimental data. Understanding these details is essential for technical teams evaluating the feasibility of integrating this route into their existing production frameworks. The answers provided reflect the specific advantages and operational parameters outlined in the intellectual property, ensuring accuracy and relevance for decision-makers.
Q: What are the primary advantages of this new condensation method over traditional Imatinib intermediate synthesis?
A: The novel method eliminates the need for complex multi-step cyclization reactions involving toxic guanidine nitrate and beta-enone precursors. Instead, it utilizes a direct, one-step acid-catalyzed coupling of commercially available substituted anilines with halopyrimidines, significantly shortening the synthetic route and improving overall yield.
Q: Which acidic catalysts are suitable for this pyrimidine coupling reaction?
A: The patent specifies a wide range of effective acidic catalysts, including inorganic acids like hydrochloric acid, sulfuric acid, and phosphoric acid, as well as organic acids such as methanesulfonic acid, p-toluenesulfonic acid, and trifluoroacetic acid, allowing for flexibility in process optimization.
Q: How does this process impact the scalability of Imatinib production?
A: By utilizing robust reaction conditions (50-100°C) and readily available starting materials, the process avoids the harsh conditions and difficult-to-source reagents of previous methods. This simplifies purification and makes the commercial scale-up of complex pharmaceutical intermediates much more feasible and economically viable.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable N-(2-methyl-5-nitro)phenyl-4-(3-pyridyl)pyrimidin-2-amine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-quality intermediates in the global fight against cancer, and we are uniquely positioned to leverage this advanced synthesis technology for our clients. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from patent to plant is seamless and efficient. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of N-phenyl-2-pyrimidinamine derivatives meets the exacting standards required for API synthesis, providing our partners with absolute confidence in material quality.
We invite pharmaceutical companies and research institutions to collaborate with us to optimize their supply chains and reduce production costs through this innovative methodology. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our capabilities can support your long-term strategic goals in oncology drug manufacturing.