Advanced Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles for Pharmaceutical Applications
Introduction to Next-Generation Triazole Synthesis
The landscape of pharmaceutical intermediate manufacturing is constantly evolving, driven by the need for more efficient, safer, and cost-effective synthetic routes. A significant breakthrough in this domain is detailed in patent CN110467579B, which discloses a novel preparation method for 5-trifluoromethyl substituted 1,2,4-triazole compounds. These heterocyclic scaffolds are ubiquitous in medicinal chemistry, serving as critical cores in numerous bioactive molecules ranging from antifungal agents like fluconazole to aromatase inhibitors like letrozole. The introduction of a trifluoromethyl group into these structures is particularly valuable, as it markedly enhances metabolic stability, lipophilicity, and bioavailability, properties that are essential for modern drug design. However, traditional methods to install this group have often been plagued by harsh conditions or expensive reagents.
This new methodology addresses these historical pain points by utilizing a metal-free, iodine-promoted cyclization strategy. By leveraging readily available starting materials such as trifluoroethylimidoyl chloride and hydrazones, the process offers a streamlined pathway to high-value intermediates. For R&D directors and procurement managers alike, understanding this technology is crucial for optimizing supply chains and reducing the cost of goods sold (COGS) in API manufacturing. The ability to synthesize these complex heterocycles without stringent anhydrous or anaerobic requirements represents a paradigm shift towards more robust and scalable chemical processes.
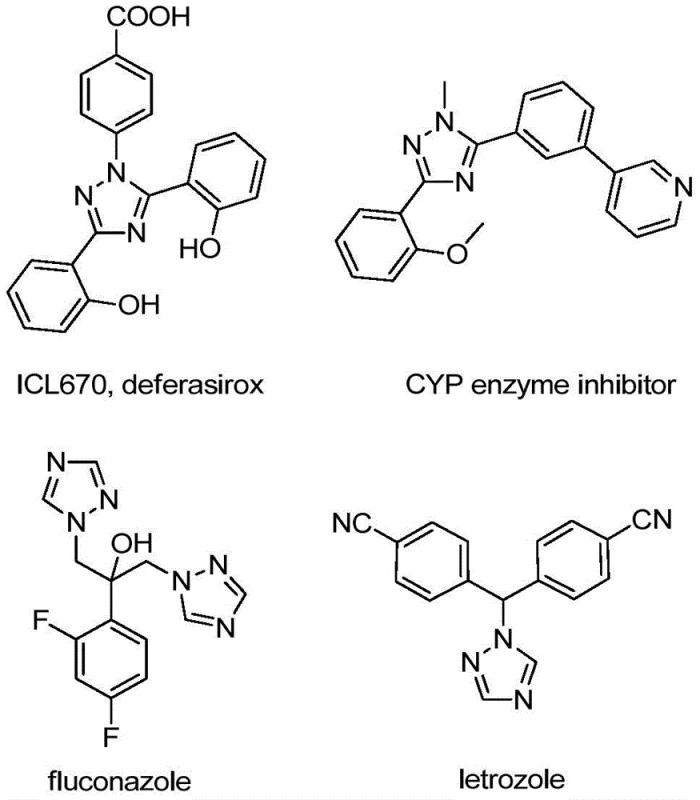
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of trifluoromethylated nitrogen-containing heterocycles has relied on two primary strategies, both of which present significant logistical and economic challenges for large-scale production. The first approach involves the direct trifluoromethylation of pre-synthesized heterocyclic rings. While conceptually straightforward, this method necessitates the use of specialized and often prohibitively expensive trifluoromethylating reagents. These reagents can be difficult to handle, sensitive to moisture, and generate substantial waste streams, complicating the environmental compliance profile of the manufacturing process. The second mainstream method employs synthons bearing the trifluoromethyl group, such as trifluorodiazoethane, reacting with coupling substrates.
Although effective in academic settings, the use of diazo compounds introduces severe safety hazards due to their potential explosivity and instability, requiring specialized equipment and rigorous safety protocols that drive up capital expenditure. Furthermore, the alternative synthon, trifluoroethylimide acid halide, has not been widely utilized in efficient cyclization reactions until recently. These conventional pathways often demand strict anhydrous and oxygen-free conditions, typically requiring gloveboxes or extensive Schlenk line techniques. Such requirements severely limit throughput and increase operational costs, making them less attractive for the commercial scale-up of complex pharmaceutical intermediates where reliability and safety are paramount.
The Novel Approach
The methodology outlined in patent CN110467579B offers a transformative solution by employing a simple, efficient, and non-metallic iodine-promoted synthesis. This novel route utilizes inexpensive and commercially available hydrazones and trifluoroethylimidoyl chloride as the foundational building blocks. The reaction proceeds smoothly in common organic solvents like dichloroethane (DCE) at moderate temperatures, specifically around 80°C. Crucially, this process eliminates the need for toxic heavy metal catalysts, which are frequently sources of contamination in pharmaceutical products and require costly removal steps to meet stringent regulatory limits.
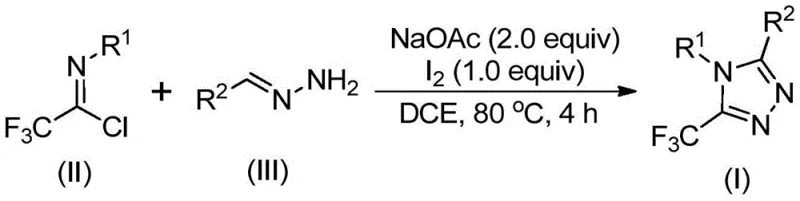
By avoiding the need for anhydrous and anaerobic conditions, this method drastically simplifies the operational workflow, allowing reactions to be conducted in standard reactor vessels without inert gas protection. The use of elemental iodine as a promoter is not only cost-effective but also facilitates a clean reaction profile. The process is highly versatile, enabling the synthesis of variously substituted 1,2,4-triazole compounds with the trifluoromethyl group positioned at different locations through rational substrate design. This flexibility widens the applicability of the method, making it a powerful tool for generating diverse libraries of drug candidates or producing specific API intermediates with high efficiency and reduced environmental impact.
Mechanistic Insights into Iodine-Promoted Cyclization
Understanding the mechanistic underpinnings of this reaction is vital for R&D teams aiming to optimize the process for specific substrates. The transformation begins with the interaction between the trifluoroethylimidoyl chloride (Structure II) and the hydrazone (Structure III) in the presence of a base, typically sodium acetate. The base promotes an intermolecular carbon-nitrogen bond formation, leading to the generation of a trifluoroacetamidine intermediate. This initial condensation step is critical and sets the stage for the subsequent ring closure. The reaction mixture is heated to facilitate this process, ensuring complete conversion of the starting materials before the introduction of the oxidant.

Following the formation of the amidine, the system undergoes isomerization, preparing the molecule for the oxidative step. The addition of elemental iodine serves as the key driver for the final aromatization. The iodine promotes an oxidative iodination, generating an iodinated intermediate that is primed for intramolecular electrophilic substitution. This cyclization event closes the five-membered triazole ring. Finally, the loss of hydrogen iodide leads to aromatization, yielding the stable 5-trifluoromethyl substituted 1,2,4-triazole product (Structure I). This mechanism highlights the dual role of the reaction conditions: the base facilitates bond formation, while the iodine drives the oxidative cyclization, ensuring a high-yielding and clean transformation without the need for transition metals.
How to Synthesize 5-Trifluoromethyl-1,2,4-Triazoles Efficiently
The practical execution of this synthesis is designed for ease of operation, making it accessible for both laboratory-scale discovery and pilot-scale production. The protocol involves a straightforward one-pot procedure where sodium acetate, trifluoroethylimidoyl chloride, and the chosen hydrazone are combined in an organic solvent. The mixture is heated to promote the initial condensation, followed by the timed addition of iodine to complete the cyclization. This simplicity reduces the training burden on operators and minimizes the risk of procedural errors. For detailed standard operating procedures and specific stoichiometric ratios optimized for different substrates, please refer to the technical guide below.
- Mix sodium acetate, trifluoroethylimidoyl chloride, and hydrazone in an organic solvent such as dichloroethane (DCE).
- Heat the reaction mixture to 80°C and stir for 2 to 4 hours to allow initial condensation and cyclization.
- Add elemental iodine to the system and continue heating for an additional 1 to 2 hours to promote oxidative aromatization, followed by standard workup and purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers tangible strategic benefits that extend beyond mere chemical yield. The primary advantage lies in the significant reduction of raw material costs. By utilizing hydrazones derived from cheap and abundant aldehydes, along with readily synthesized trifluoroethylimidoyl chloride, the bill of materials is kept low. Furthermore, the elimination of expensive noble metal catalysts removes a major cost center associated with both the purchase of the catalyst and the downstream processing required to remove trace metal residues to ppm levels.
- Cost Reduction in Manufacturing: The process achieves cost efficiency through multiple vectors. Firstly, the avoidance of hazardous reagents like trifluorodiazoethane eliminates the need for specialized safety infrastructure and insurance costs associated with explosive materials. Secondly, the use of elemental iodine and sodium acetate as promoters is economically superior to transition metal systems. Thirdly, the simplified workup procedure, which often involves basic filtration and chromatography, reduces solvent consumption and labor hours. These factors collectively contribute to a lower cost per kilogram of the final intermediate, enhancing the margin potential for the final API.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the use of commodity chemicals. The starting materials, including various substituted aromatic amines and aldehydes, are widely available from multiple global suppliers, reducing the risk of single-source bottlenecks. The robustness of the reaction conditions—specifically the tolerance to moisture and oxygen—means that production is less susceptible to delays caused by equipment failure or environmental control issues. This reliability ensures consistent delivery schedules, a critical metric for maintaining continuous API manufacturing lines.
- Scalability and Environmental Compliance: From an EHS (Environment, Health, and Safety) perspective, this method is highly favorable. The absence of heavy metals simplifies waste treatment and disposal, aligning with increasingly strict environmental regulations. The reaction can be easily scaled from gram to kilogram quantities without significant re-optimization, as demonstrated by the patent's successful expansion to gram-level synthesis. The use of standard solvents like DCE allows for easy recovery and recycling, further minimizing the environmental footprint and supporting sustainable manufacturing practices.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and specifications provided in the patent literature, ensuring accuracy and relevance for technical decision-makers. Understanding these details helps in assessing the feasibility of integrating this route into existing production workflows.
Q: What are the key advantages of this iodine-promoted method over traditional trifluoromethylation?
A: Unlike traditional methods that often require expensive trifluoromethylating reagents or unstable diazo compounds, this method utilizes cheap and stable trifluoroethylimidoyl chloride. Furthermore, it operates under mild conditions without the need for strict anhydrous or anaerobic environments, significantly simplifying operational complexity.
Q: Does this synthesis require heavy metal catalysts?
A: No, one of the distinct advantages of the process described in patent CN110467579B is that it avoids the use of toxic heavy metal catalysts. It relies on elemental iodine and sodium acetate, which facilitates easier downstream purification and reduces environmental hazards associated with metal waste.
Q: What is the substrate scope for this reaction?
A: The method demonstrates broad substrate tolerance. It accommodates various substituted aryl groups on both the imidoyl chloride and the hydrazone components, including electron-donating groups like methyl and methoxy, as well as electron-withdrawing groups like halogens and nitro groups, allowing for the synthesis of diverse triazole derivatives.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethyl-1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the drug development lifecycle. Our team of expert chemists has extensively evaluated the technology described in patent CN110467579B and confirmed its potential for robust commercial application. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition seamlessly from clinical trials to market launch. Our facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 5-trifluoromethyl-1,2,4-triazole intermediate meets the highest industry standards.
We invite you to collaborate with us to leverage this advanced synthetic route for your next project. By partnering with our technical procurement team, you can obtain a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to reach out today to request specific COA data and route feasibility assessments, allowing us to demonstrate how our expertise can drive efficiency and reliability in your supply chain.