Advanced Pd-Catalyzed Synthesis of Trelagliptin Intermediates for Commercial Scale-Up
The pharmaceutical landscape for Type 2 diabetes treatment continues to evolve, with long-acting DPP-IV inhibitors like Trelagliptin representing a critical therapeutic class. A recent technological breakthrough, documented in patent CN108794448B, introduces a robust preparation method for Trelagliptin and its salts that addresses longstanding manufacturing bottlenecks. This innovation shifts the paradigm from traditional nucleophilic substitution to a sophisticated palladium-catalyzed cross-coupling strategy. By leveraging advanced organometallic chemistry, the process effectively suppresses the formation of structurally similar impurities that have historically plagued production lines. For global supply chain stakeholders, this represents a pivotal opportunity to secure a reliable trelagliptin intermediate supplier capable of delivering high-purity materials with consistent quality. The structural integrity of the final active pharmaceutical ingredient is paramount, and this method ensures that the delicate balance of functional groups is maintained throughout the synthesis.
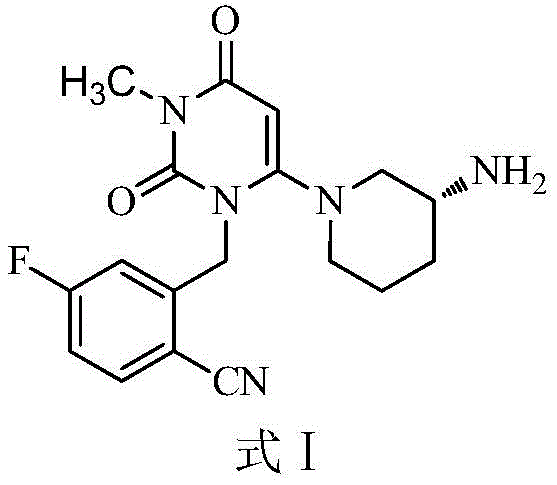
Understanding the molecular architecture is the first step toward appreciating the synthetic challenges involved. As illustrated in the chemical structure, Trelagliptin features a complex arrangement including a pyrimidine-dione core linked to a fluorobenzonitrile group and a chiral piperidine ring. The presence of both a chlorine atom on the pyrimidine ring and a fluorine atom on the benzene ring creates a unique reactivity profile. In conventional synthesis attempts, differentiating between these two halogenated sites is difficult, often leading to non-selective reactions. The new methodology described in the patent navigates this complexity by employing a protected amine precursor, which acts as a steric and electronic gatekeeper, ensuring that the coupling reaction occurs precisely where intended. This level of precision is essential for meeting the stringent regulatory standards required for high-purity pharmaceutical intermediates in the global market.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the preparation of Trelagliptin has relied on direct nucleophilic substitution reactions using (R)-3-aminopiperidine dihydrochloride. While conceptually straightforward, this approach suffers from severe chemoselectivity issues due to the electronic nature of the substrate. The benzene ring in the starting material possesses a fluorine atom at the para position relative to a strongly electron-withdrawing cyano group. This configuration renders the fluorine atom unusually reactive, transforming it into a viable leaving group under basic conditions. Consequently, the amine nucleophile attacks not only the intended chlorine site on the pyrimidine ring but also the fluorine site on the benzene ring. This side reaction generates a bis-substituted byproduct, identified as Impurity 5, which can constitute up to 12% of the crude reaction mixture.
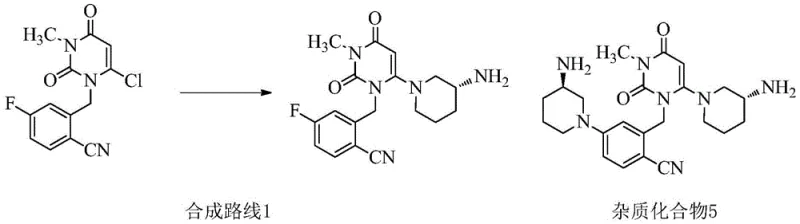
The presence of Impurity 5 poses a significant downstream processing challenge because its physicochemical properties are remarkably similar to those of the target Trelagliptin molecule. Separating these closely related compounds typically requires resource-intensive techniques such as High Performance Liquid Chromatography (HPLC) or extensive column chromatography. These purification steps are not only costly but also difficult to scale, leading to prolonged production cycles and reduced overall throughput. In comparative test examples provided in the patent data, the conventional method yielded only 63.2% of the target product with a purity of 98.8%, necessitating complex workups that are ill-suited for multi-ton commercial manufacturing. This inefficiency highlights the urgent need for cost reduction in API manufacturing through smarter synthetic design.
The Novel Approach
The innovative route disclosed in patent CN108794448B circumvents these pitfalls by introducing a protecting group strategy coupled with transition metal catalysis. Instead of using the free amine, the process utilizes 3-tert-butyloxycarbonyl-aminopiperidine (Compound 7). The tert-butyloxycarbonyl (Boc) group serves a dual purpose: it reduces the nucleophilicity of the nitrogen atom just enough to prevent attack on the less reactive aryl fluoride, while still allowing reaction at the more activated chloro-pyrimidine site under palladium catalysis. This selectivity is the cornerstone of the new method, effectively eliminating the formation of Impurity 5 at the source rather than trying to remove it later. The reaction proceeds smoothly in polar aprotic solvents like DMSO, utilizing a palladium catalyst system that facilitates the C-N bond formation with high efficiency.
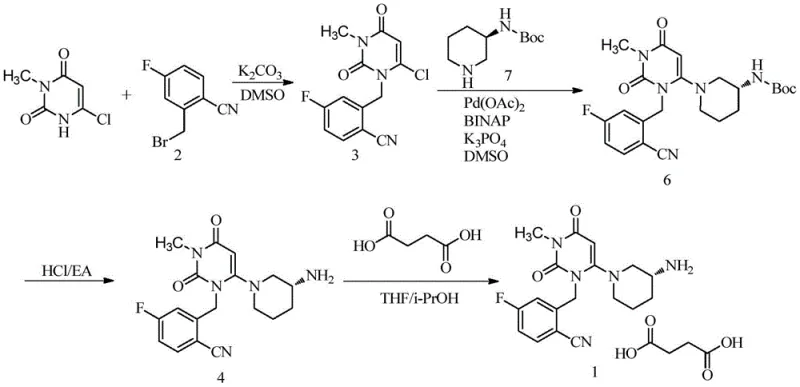
Furthermore, the novel approach simplifies the isolation and purification stages significantly. Following the coupling reaction, the intermediate (Compound 6) can be isolated through standard aqueous workup and extraction, avoiding the need for chromatographic separation. The subsequent deprotection step uses hydrochloric acid in ethyl acetate, precipitating the product as a solid which can be easily filtered and washed. This streamlined workflow translates directly into operational excellence, enabling the commercial scale-up of complex pharmaceutical intermediates with minimal waste generation. The patent reports yields exceeding 95% and purities greater than 99%, demonstrating that this method is not just theoretically sound but practically superior for industrial application. By removing the bottleneck of difficult purification, manufacturers can achieve substantial improvements in production velocity and cost efficiency.
Mechanistic Insights into Pd-Catalyzed C-N Cross-Coupling
The core of this technological advancement lies in the palladium-catalyzed amination reaction, a variation of the Buchwald-Hartwig coupling. The mechanism initiates with the oxidative addition of the palladium(0) species into the carbon-chlorine bond of the pyrimidine ring (Compound 3). This step is facilitated by the electron-deficient nature of the heterocycle, which makes the C-Cl bond susceptible to metal insertion. The choice of ligand is critical here; bulky, electron-rich phosphine ligands such as BINAP, DPPF, or X-PHOS stabilize the palladium center and accelerate the oxidative addition. Once the Pd(II) intermediate is formed, the protected piperidine coordinates to the metal center. In the presence of a base like potassium phosphate (K3PO4), the amine is deprotonated, forming a palladium-amido complex. This complex then undergoes reductive elimination to forge the new C-N bond, releasing the coupled product (Compound 6) and regenerating the active Pd(0) catalyst for the next cycle.
Controlling the impurity profile is inherently built into this mechanistic pathway. Because the reaction relies on the specific activation of the C-Cl bond by the palladium catalyst, the C-F bond on the benzene ring remains inert under these conditions. The palladium catalyst does not facilitate oxidative addition into the aryl-fluorine bond in this specific substrate context, especially when the milder, protected amine is used. This orthogonal reactivity ensures that the fluorine atom remains untouched, preserving the structural integrity required for biological activity. Additionally, the use of the Boc group prevents over-alkylation or polymerization side reactions that free amines might undergo. The result is a clean reaction profile where the primary output is the desired intermediate, drastically reducing the burden on quality control laboratories and ensuring that the final high-purity trelagliptin meets all pharmacopeial specifications without extensive reprocessing.
How to Synthesize Trelagliptin Efficiently
Implementing this synthesis requires precise control over reaction parameters to maximize the benefits of the catalytic system. The process begins with the preparation of the chloro-pyrimidine precursor (Compound 3) via alkylation, followed by the crucial palladium-catalyzed step. Operators must maintain an inert nitrogen atmosphere to protect the catalyst from oxidation and carefully control the temperature between 40°C and 100°C, with 80°C being optimal for balancing reaction rate and selectivity. The stoichiometry of the reagents is also vital; a slight excess of the protected amine and base ensures complete conversion of the starting material. Detailed standard operating procedures for each stage, including solvent ratios and workup pH adjustments, are essential for reproducibility. For a comprehensive breakdown of the specific experimental conditions and reagent quantities, please refer to the standardized synthesis guide below.
- Perform N-alkylation of 6-chloro-3-methyluracil with 2-cyano-5-fluorobenzyl bromide using potassium carbonate in DMSO to obtain Compound 3.
- Execute a Pd-catalyzed cross-coupling reaction between Compound 3 and 3-Boc-aminopiperidine using Pd(OAc)2, a phosphine ligand (e.g., BINAP), and K3PO4 in DMSO at 80°C to yield Compound 6.
- Deprotect Compound 6 using HCl in ethyl acetate, followed by pH adjustment and extraction to isolate pure Trelagliptin (Compound 4), optionally forming the succinate salt.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented methodology offers transformative economic and logistical benefits. The primary driver of value is the drastic simplification of the purification process. By eliminating the need for preparative HPLC or column chromatography, manufacturers can significantly reduce the consumption of expensive solvents and silica gel, which are major cost centers in fine chemical production. This reduction in material usage directly correlates to a lower cost of goods sold (COGS), allowing for more competitive pricing in the tender market. Moreover, the simplified workflow reduces the time required for batch processing, effectively increasing the capacity of existing manufacturing facilities without the need for capital expenditure on new equipment. This efficiency gain is crucial for meeting the growing global demand for diabetes medications.
- Cost Reduction in Manufacturing: The elimination of complex chromatographic purification steps results in substantial savings on consumables and labor. Traditional methods require significant amounts of eluents and stationary phases to separate Impurity 5, whereas this new route produces a crude product of such high purity that simple crystallization or extraction suffices. Additionally, the high yield (>95%) means that less raw material is wasted, optimizing the atom economy of the process. The removal of transition metal residues is also streamlined, as the catalyst loading is low and the workup is designed to sequester palladium effectively, reducing the cost associated with metal scavenging resins.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route enhances supply security by reducing the risk of batch failures. Conventional methods are sensitive to minor variations in reaction conditions, which can lead to spikes in impurity levels and rejected batches. In contrast, the Pd-catalyzed method is tolerant and reproducible, ensuring a steady flow of material. Furthermore, the raw materials required, such as the Boc-protected piperidine and standard palladium ligands, are commercially available from multiple global vendors, mitigating the risk of single-source dependency. This diversification of the supply base ensures reducing lead time for high-purity intermediates and maintains continuity of supply even during market fluctuations.
- Scalability and Environmental Compliance: From an environmental perspective, this process aligns with green chemistry principles by minimizing waste generation. The avoidance of large volumes of chromatography solvents reduces the facility's volatile organic compound (VOC) emissions and hazardous waste disposal costs. The use of DMSO as a solvent, while requiring careful recovery, is manageable on a large scale and allows for high concentration reactions, further reducing the solvent footprint. The ability to scale this process from kilogram to multi-ton quantities without changing the fundamental chemistry makes it an ideal candidate for long-term commercial production, ensuring that environmental compliance is maintained alongside economic growth.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and claims within patent CN108794448B, providing a factual basis for decision-making. Understanding these nuances helps R&D and procurement teams evaluate the feasibility of adopting this route for their specific manufacturing needs. The answers cover aspects ranging from catalyst selection to impurity management, offering a holistic view of the process capabilities.
Q: Why is the Boc-protected piperidine used instead of the free amine in this synthesis?
A: The use of 3-tert-butyloxycarbonyl-aminopiperidine (Boc-protected) is critical for chemoselectivity. In conventional methods using the free amine, the strong electron-withdrawing cyano group activates the fluorine atom on the benzene ring, leading to unwanted bis-substitution (Impurity 5). The Boc group modulates the nucleophilicity, ensuring reaction occurs exclusively at the chlorine position on the pyrimidine ring, thereby drastically reducing impurity formation.
Q: What represents the key advantage of this Pd-catalyzed route over traditional nucleophilic substitution?
A: The primary advantage is the significant improvement in yield and purity without complex purification. Traditional routes often yield crude products containing up to 12% of difficult-to-remove impurities, requiring HPLC purification and resulting in yields around 63%. This novel Pd-catalyzed method achieves yields exceeding 95% with purity greater than 99%, utilizing simple filtration and extraction workups suitable for industrial scale-up.
Q: Which ligands are effective for this specific palladium-catalyzed amination?
A: The patent specifies several effective bidentate and monodentate phosphine ligands that facilitate the coupling under mild conditions. These include BINAP ((±)-2,2'-bis(diphenylphosphino)-1,1'-binaphthyl), DPPF (1,1'-bis(diphenylphosphino)ferrocene), tri-tert-butylphosphine, X-PHOS, and S-PHOS. These ligands stabilize the palladium catalyst and promote the oxidative addition and reductive elimination steps essential for high conversion.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trelagliptin Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthesis routes in the modern pharmaceutical industry. Our team of expert chemists has extensively analyzed the technology described in CN108794448B and is fully equipped to implement this advanced Pd-catalyzed methodology. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from lab-scale optimization to full-scale manufacturing is seamless. Our state-of-the-art facilities are designed to handle sensitive organometallic reactions with precision, and our rigorous QC labs enforce stringent purity specifications to guarantee that every batch of Trelagliptin intermediate meets the highest international standards. We are committed to delivering value through technical excellence and operational reliability.
We invite potential partners to engage with us to explore how this optimized synthesis route can benefit your supply chain. By leveraging our expertise, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us collaborate to drive down costs and enhance the availability of life-saving diabetes treatments globally, ensuring a secure and efficient supply of high-quality Trelagliptin intermediates for your organization.