Advanced Stereoselective Synthesis of Z-Fluoro-Difluoromethylene Olefins for Commercial API Manufacturing
Introduction to Next-Generation Fluorinated Olefin Synthesis
The integration of fluorine atoms into organic scaffolds remains a cornerstone strategy in modern medicinal chemistry, profoundly influencing the metabolic stability and lipophilicity of drug candidates. Patent CN111517954A introduces a groundbreaking methodology for the stereoselective synthesis of (Z)-5-fluoro-2-difluoromethylene olefin derivatives, addressing a long-standing challenge in constructing thermodynamically unstable Z-alkenes. This technology leverages a sophisticated radical cascade mechanism to achieve remote C(sp3)-H fluorination, bypassing the limitations of traditional transition-metal catalysis that often require cumbersome directing groups. For R&D directors and procurement specialists, this represents a significant leap forward in accessing high-value fluorinated building blocks essential for next-generation API development. The ability to install both fluorine and difluoromethylene moieties in a single operational step drastically simplifies the synthetic landscape, offering a robust pathway for the reliable pharmaceutical intermediate supplier market.
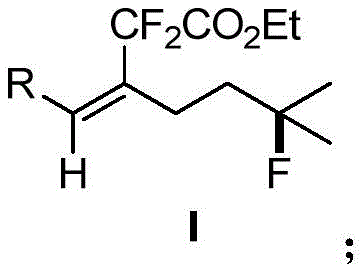
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the functionalization of inert C-H bonds has relied heavily on transition metal catalysis assisted by directing groups, a strategy that inherently increases step count and reduces overall atom economy. Conventional approaches to synthesizing fluorinated olefins often struggle with regioselectivity and stereoselectivity, predominantly yielding the thermodynamically stable (E)-isomers rather than the biologically relevant (Z)-configurations. Furthermore, the requirement for pre-functionalized substrates or harsh reaction conditions limits the scope of compatible functional groups, creating bottlenecks in the synthesis of complex drug molecules. These inefficiencies translate directly into higher manufacturing costs and extended lead times, posing significant challenges for cost reduction in API manufacturing where streamlined processes are critical for commercial viability.
The Novel Approach
The methodology disclosed in the patent utilizes a silver-mediated radical cascade that initiates with the addition of a difluoromethylene radical to an internal alkyne, triggering a subsequent 1,5-hydrogen migration. This innovative sequence allows for the direct transformation of simple internal alkynes into complex (Z)-fluorinated products without the need for pre-installed directing groups. By operating under mild conditions at 25°C in acetonitrile, this process demonstrates exceptional functional group tolerance, accommodating diverse substituents such as halogens, esters, and nitriles. This shift from multi-step directed metallation to a one-pot radical functionalization represents a paradigm shift, enabling the efficient construction of multiple chemical bonds including C(sp2)-CF2 and C(sp3)-F simultaneously, thereby enhancing the overall efficiency of fine chemical intermediate production.
Mechanistic Insights into Silver-Mediated Radical Cascade Fluorination
The core of this technological breakthrough lies in the precise orchestration of radical species to achieve remote functionalization with high stereocontrol. The reaction initiates with the generation of a difluoromethylene radical from ethyl trimethylsilyl difluoroacetate (TMSCF2CO2Et) mediated by the AgNO3/CsF/PhI(OCOCF3)2 system. This radical adds to the internal alkyne to form a vinyl radical intermediate, which is pivotal for the subsequent intramolecular 1,5-hydrogen atom transfer (HAT). This HAT step generates a distal carbon-centered radical at the C(sp3) position, which is then trapped by the fluorine source, Selectfluor, to finalize the C-F bond formation. Understanding this mechanistic pathway is crucial for R&D teams aiming to optimize reaction parameters for specific substrate classes.
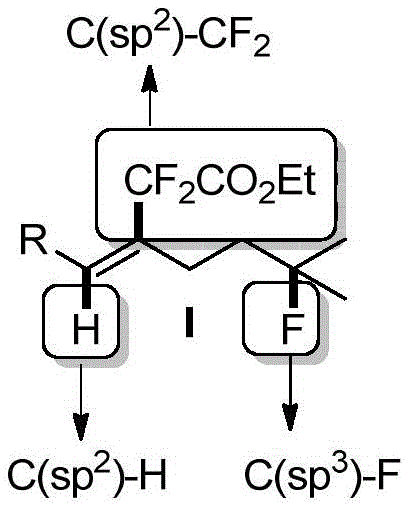
Impurity control in this radical system is managed through the inherent selectivity of the 1,5-hydrogen migration, which favors the formation of a specific six-membered transition state, minimizing side reactions such as cyclization that often plague radical cascades. The use of Selectfluor as the terminal oxidant and fluorine source ensures that the radical intermediate is quenched efficiently to form the desired C-F bond rather than undergoing dimerization or hydrogen abstraction from the solvent. This high level of chemoselectivity ensures that the final product profile is clean, reducing the burden on downstream purification processes. For quality assurance teams, this mechanistic robustness translates to consistent batch-to-batch reproducibility and stringent purity specifications, which are non-negotiable requirements for supplying active pharmaceutical ingredients.
How to Synthesize (Z)-5-fluoro-2-difluoromethylene olefin Efficiently
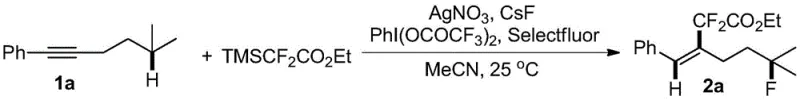
The practical implementation of this synthesis involves a straightforward one-pot procedure that combines the alkyne substrate with the fluorinating reagents in a polar aprotic solvent. The reaction is conducted under a nitrogen atmosphere to prevent radical quenching by oxygen, ensuring maximum yield and conversion. Following the reaction period, a standard aqueous workup involving extraction and silica gel chromatography affords the pure product. This operational simplicity makes the process highly attractive for rapid prototyping in process chemistry labs. The detailed standardized synthesis steps are outlined in the guide below, providing a clear roadmap for technical teams to replicate these results.
- Mix internal alkyne, Selectfluor, TMSCF2CO2Et, AgNO3, CsF, and PhI(OCOCF3)2 in acetonitrile solvent.
- Stir the reaction mixture at room temperature (25°C) for 18 hours under nitrogen atmosphere.
- Quench with water, extract with ethyl acetate, wash with brine, dry over sodium sulfate, and purify via silica gel chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain perspective, the adoption of this radical fluorination technology offers substantial strategic benefits by simplifying the raw material portfolio and reducing dependency on exotic catalysts. The elimination of expensive transition metal catalysts like palladium or rhodium, which often require rigorous removal steps to meet residual metal limits in APIs, leads to significant process intensification. This streamlining not only reduces the consumption of precious metals but also minimizes the generation of hazardous waste streams associated with metal scavenging, aligning with modern green chemistry principles and environmental compliance standards. Consequently, this contributes to substantial cost savings in the overall manufacturing budget without compromising on product quality or safety.
- Cost Reduction in Manufacturing: The one-step nature of this transformation eliminates the need for multiple isolation and purification stages typically required in stepwise functionalization strategies. By avoiding the synthesis and subsequent removal of directing groups, the process reduces solvent usage, labor hours, and energy consumption associated with extended reaction times and heating cycles. This consolidation of synthetic steps directly lowers the cost of goods sold (COGS), making the production of high-purity pharmaceutical intermediates more economically viable for large-scale operations.
- Enhanced Supply Chain Reliability: The reagents utilized in this protocol, including silver nitrate, cesium fluoride, and Selectfluor, are commercially available commodities with stable supply chains, mitigating the risk of raw material shortages. Unlike specialized ligands or custom-synthesized catalysts that may have long lead times, these bulk chemicals ensure consistent availability for continuous manufacturing campaigns. This reliability is critical for maintaining uninterrupted production schedules and meeting the just-in-time delivery expectations of global pharmaceutical clients.
- Scalability and Environmental Compliance: The reaction proceeds at ambient temperature (25°C), removing the need for energy-intensive heating or cryogenic cooling systems, which simplifies reactor design and enhances process safety during scale-up. The mild conditions reduce the thermal load on manufacturing facilities and lower the risk of runaway reactions, facilitating the commercial scale-up of complex fluorinated compounds. Furthermore, the simplified workup procedure reduces the volume of organic waste generated, supporting sustainability goals and reducing disposal costs associated with hazardous chemical waste management.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this fluorination technology. These insights are derived directly from the experimental data and mechanistic studies presented in the patent literature, providing clarity on reaction performance and substrate compatibility. Understanding these nuances helps technical procurement teams evaluate the feasibility of integrating this method into existing production workflows.
Q: What is the primary advantage of this radical method over traditional transition metal catalysis?
A: This method eliminates the need for pre-installed directing groups, significantly shortening the synthetic route and avoiding the costly removal of heavy metal residues often associated with traditional C-H activation strategies.
Q: How does this process achieve stereoselectivity for the thermodynamically unstable (Z)-isomer?
A: The reaction proceeds through a unique vinyl radical intermediate induced by alkyne radical addition, which facilitates a trans-hydrogen-radical addition pathway, effectively overcoming the thermodynamic preference for the (E)-isomer found in conventional methods.
Q: Is this synthesis scalable for industrial production of pharmaceutical intermediates?
A: Yes, the reaction operates under mild conditions (25°C) without requiring extreme pressures or temperatures, and utilizes commercially available reagents, making it highly suitable for commercial scale-up of complex pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable (Z)-5-fluoro-2-difluoromethylene olefin Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced fluorination technologies play in accelerating drug discovery and development pipelines. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative laboratory methods like the one described in CN111517954A can be successfully translated into robust industrial processes. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of (Z)-5-fluoro-2-difluoromethylene olefin meets the highest international standards for pharmaceutical applications.
We invite you to collaborate with us to leverage this cutting-edge synthesis for your next project. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific molecule. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our expertise can optimize your supply chain and reduce time-to-market for your fluorinated drug candidates.