Scalable Total Synthesis of Anti-Inflammatory Alkaloid Arbornamine for Commercial API Production
Scalable Total Synthesis of Anti-Inflammatory Alkaloid Arbornamine for Commercial API Production
The pharmaceutical industry is constantly seeking efficient pathways to access complex natural products that possess significant therapeutic potential. A recent breakthrough detailed in patent CN113698402B outlines a robust synthetic method for Arbornamine, a pentacyclic monoterpene indole alkaloid originally isolated from Pistia plants. This molecule is characterized by a unique structural framework containing three chiral centers, including a challenging aza quaternary carbon center, which has historically posed significant hurdles for total synthesis. The disclosed method not only overcomes these stereochemical challenges but also demonstrates that the synthetic product retains anti-inflammatory activity equivalent to the standard drug indomethacin. For R&D teams and procurement specialists, this represents a pivotal shift from scarce natural extraction to reliable chemical manufacturing. 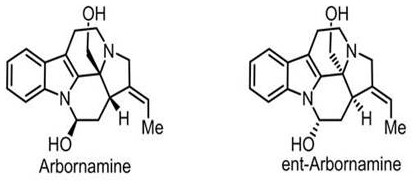
As a reliable pharmaceutical intermediate supplier, understanding the nuances of such synthetic routes is critical for ensuring supply chain continuity. The patent describes a comprehensive eight-step sequence starting from commercially available monobenzyl-protected tryptamine hydrochloride. By leveraging well-established reactions such as the Pictet-Spengler cyclization and the Heck coupling reaction, the inventors have created a pathway that is amenable to scale-up. This transition from laboratory curiosity to industrial feasibility is essential for companies looking to develop novel anti-inflammatory agents without being bottlenecked by the low yields and high costs associated with plant extraction.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this innovation, the total synthesis of Arbornamine was a formidable task, with the first reported route by the Yang group in 2018 serving as a primary benchmark. While scientifically elegant, that earlier approach relied on a nickel-catalyzed reductive Heck reaction to construct the pentacyclic skeleton, a process that often demands rigorous exclusion of air and moisture and specialized catalysts that can drive up operational costs. Furthermore, the conventional route involved the synthesis of a tetracyclic lactam intermediate followed by complex functional group conversions, which introduced multiple purification steps and potential yield losses. 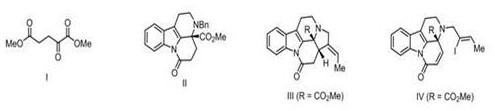
From a manufacturing perspective, the reliance on strong acids and high temperatures in some traditional indole alkaloid syntheses can lead to poor functional group compatibility, resulting in the formation of difficult-to-remove impurities. These impurities complicate downstream processing and increase the burden on quality control laboratories. Additionally, the scarcity of specific precursors required for older routes often creates supply chain vulnerabilities. For a procurement manager, these factors translate into higher risk profiles and unpredictable lead times, making the adoption of such legacy methods less attractive for commercial-scale production of high-purity pharmaceutical intermediates.
The Novel Approach
The methodology presented in CN113698402B offers a streamlined alternative that prioritizes operational simplicity and cost reduction in pharmaceutical intermediate manufacturing. The new route initiates with a Pictet-Spengler cyclization between a tryptamine derivative and diethyl ketomalonate, a reaction known for its atom economy and reliability in forming tetrahydro-beta-carboline scaffolds. This is followed by a reduction to a diol and a strategic selective protection step that efficiently constructs the crucial chiral quaternary carbon center without requiring exotic chiral auxiliaries. 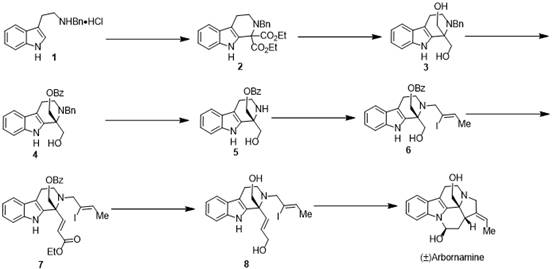
Subsequent steps involve N-alkylation to introduce an iodoolefin side chain, followed by oxidation and a Horner-Wadsworth-Emmons (HWE) reaction to set up the final ring closure. The process culminates in an intramolecular Heck coupling, which elegantly closes the pentacyclic ring system under relatively mild conditions compared to previous nickel-catalyzed variants. This approach significantly simplifies the synthetic tree, reducing the number of isolation steps and improving the overall throughput. For supply chain heads, this translates to a more robust process with better scalability, ensuring that the commercial scale-up of complex alkaloids can be achieved with greater predictability and lower environmental impact.
Mechanistic Insights into Pictet-Spengler Cyclization and Heck Coupling
The success of this synthetic strategy hinges on the precise execution of two key mechanistic transformations: the initial Pictet-Spengler cyclization and the final Heck coupling. The Pictet-Spengler reaction involves the condensation of the tryptamine amine with the ketone of the diester, followed by an electrophilic aromatic substitution that forms the new C-C bond at the indole C2 position. This step is critical as it establishes the core tetracyclic framework upon which the rest of the molecule is built. The use of diethyl ketomalonate is particularly advantageous as it introduces the necessary ester functionality for subsequent reduction, while the acidic conditions promote the cyclization with high regioselectivity, minimizing the formation of regioisomeric byproducts that could compromise purity.
The final intramolecular Heck coupling is the masterpiece of this synthesis, forging the bond that completes the pentacyclic architecture. Mechanistically, this involves the oxidative addition of a palladium catalyst to the vinyl iodide moiety installed in earlier steps, followed by migratory insertion into the pendant alkene and beta-hydride elimination to restore the double bond. The patent specifies the use of palladium acetate with triphenylphosphine ligands, a classic catalytic system that offers a balance of reactivity and stability. This choice of catalyst system is vital for impurity control, as it avoids the over-reduction or isomerization side reactions often seen with more aggressive metal catalysts, thereby ensuring the final product meets stringent purity specifications required for biological testing and potential clinical applications.
How to Synthesize Arbornamine Efficiently
Implementing this synthesis requires careful attention to reaction conditions, particularly during the reduction and protection steps where stereochemistry is established. The process begins with the preparation of the diester intermediate, followed by reduction to the diol using lithium aluminum hydride, a powerful reductant that must be handled with care to ensure safety and reproducibility. The subsequent selective protection of the hydroxyl group is the key to unlocking the correct stereochemical outcome, creating the quaternary center that defines the molecule's 3D shape. For detailed operational parameters, stoichiometry, and workup procedures, please refer to the standardized protocol below.
- Perform Pictet-Spengler cyclization on monobenzyl-protected tryptamine hydrochloride with diethyl ketomalonate to form the diester intermediate.
- Reduce the diester to a diol using lithium aluminum hydride, followed by selective hydroxyl protection to construct the chiral quaternary carbon center.
- Execute N-alkylation with iodoolefin, followed by oxidation and HWE reaction to install the necessary side chain for final cyclization.
- Complete the synthesis via intramolecular Heck coupling reaction to close the pentacyclic skeleton and yield Arbornamine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the shift from extraction to this specific synthetic route offers profound strategic benefits. The primary advantage lies in the decoupling of production from agricultural variables; unlike plant extraction, which is subject to seasonal fluctuations, climate change, and geopolitical instability in sourcing regions, chemical synthesis provides a consistent, year-round supply of the active ingredient. The starting materials, such as tryptamine derivatives and simple diesters, are commodity chemicals available from multiple global vendors, which mitigates the risk of single-source dependency and enhances supply chain resilience significantly.
- Cost Reduction in Manufacturing: The economic viability of this route is driven by the use of inexpensive, commercially available raw materials and the avoidance of precious metal catalysts like nickel in favor of more standard palladium systems that can be recovered and recycled. By eliminating the need for complex chiral resolution steps early in the synthesis and relying on diastereoselective transformations, the process reduces the overall consumption of solvents and reagents. This streamlined approach lowers the cost of goods sold (COGS), allowing for more competitive pricing in the final API market while maintaining healthy margins for manufacturers.
- Enhanced Supply Chain Reliability: The synthetic pathway is designed with robustness in mind, utilizing reactions that are tolerant to minor variations in temperature and mixing, which is crucial for technology transfer from lab to pilot plant. The functional group compatibility mentioned in the patent implies that the process is less prone to failure due to trace impurities in feedstocks, a common cause of batch failures in fine chemical manufacturing. This reliability ensures that delivery schedules can be met consistently, reducing the need for safety stock and freeing up working capital for our partners.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, the route avoids the use of highly toxic reagents or extreme conditions that would require specialized containment infrastructure. The waste streams generated are primarily organic solvents and salt byproducts, which are manageable through standard wastewater treatment protocols. This ease of waste management facilitates faster regulatory approval for new manufacturing sites and supports the industry's broader goals of green chemistry, making it an attractive option for companies with strict sustainability mandates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of Arbornamine. These answers are derived directly from the technical disclosures in patent CN113698402B and general industry knowledge regarding alkaloid synthesis. Understanding these details helps stakeholders make informed decisions about integrating this intermediate into their development pipelines.
Q: What is the biological activity profile of synthetically produced Arbornamine?
A: According to patent CN113698402B, the synthesized Arbornamine exhibits anti-inflammatory activity equivalent to that of indomethacin, making it a potent candidate for further drug development.
Q: How does this new synthesis method improve upon previous routes?
A: Unlike earlier methods requiring harsh conditions or complex nickel-catalyzed steps, this route utilizes mild Pictet-Spengler cyclization and standard palladium-catalyzed Heck coupling, offering better functional group compatibility and lower raw material costs.
Q: Is the construction of the chiral quaternary carbon center challenging in this process?
A: The process addresses this key challenge through a selective hydroxyl protection strategy on the diol intermediate, efficiently constructing the critical aza quaternary carbon chiral center required for the pentacyclic framework.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Arbornamine Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of efficient synthetic routes like the one described for Arbornamine. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can move seamlessly from gram-scale research to multi-ton manufacturing. Our facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of Arbornamine or related intermediate performs consistently in your biological assays and formulation studies.
We invite you to collaborate with us to leverage this advanced synthesis technology for your anti-inflammatory drug development programs. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how our optimized processes can reduce your overall R&D expenditure. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us help you secure a stable, high-quality supply of this valuable natural product analogue.