Scalable Synthesis of 3-Fluoroazetidine Derivatives for High-Purity API Manufacturing
Scalable Synthesis of 3-Fluoroazetidine Derivatives for High-Purity API Manufacturing
The rapidly evolving landscape of pharmaceutical discovery increasingly relies on fluorinated heterocycles to modulate metabolic stability and bioavailability. A pivotal development in this domain is detailed in patent CN102731362B, which discloses a highly efficient method for preparing 1-carboxylic acid tert-butyl ester-3-fluoro-azetidine derivatives. This specific class of compounds serves as a critical building block for various therapeutic agents, including dipeptidyl peptidase-IV (DPP-IV) inhibitors and cannabinoid receptor modulators. The technical breakthrough presented in this patent addresses the longstanding challenges associated with introducing fluorine atoms into strained four-membered rings, offering a pathway that drastically reduces synthetic complexity while maximizing output. For R&D directors and procurement strategists, understanding this methodology is essential for securing a reliable supply chain of high-purity pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 3-fluoroazetidine derivatives has been plagued by inefficient multi-step sequences that hinder commercial viability. As illustrated in the prior art referenced within the patent documentation, traditional routes often necessitate up to eight distinct reaction steps to reach the target molecule. These legacy pathways typically involve harsh reagents such as N-bromosuccinimide (NBS) and transition metal catalysts like Ruthenium chloride, which introduce significant impurities and complicate downstream purification. Furthermore, the cumulative effect of performing eight sequential transformations results in abysmal overall yields, often reported as low as 2.46%. Such inefficiencies not only drive up the cost of goods sold but also create substantial bottlenecks in the supply chain, making it difficult to source these materials in the quantities required for clinical trials or commercial launch.
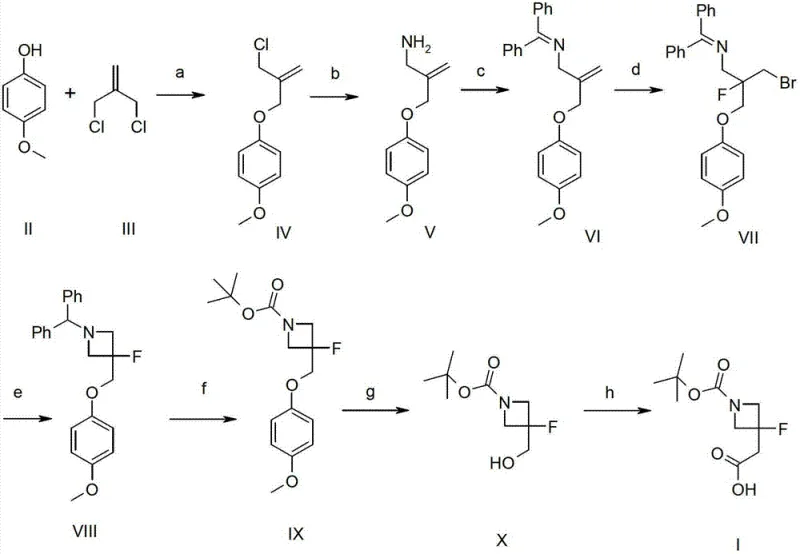
The Novel Approach
In stark contrast to the cumbersome historical methods, the invention described in CN102731362B introduces a streamlined three-step strategy that fundamentally reshapes the production economics of this intermediate. The new route begins with the protection of azetidine-3-carboxylic acid methyl ester, followed by a direct electrophilic fluorination using N-fluorobenzenesulfonimide (NFSI), and concludes with a simple hydrolysis. This approach eliminates the need for toxic heavy metals and reduces the operational burden on manufacturing teams. By condensing the synthesis into just three high-yielding steps, the process achieves a remarkable total yield of 74% for the fluorinated intermediate and nearly quantitative conversion (99%) in the final hydrolysis step. This dramatic improvement in efficiency translates directly into cost reduction in pharmaceutical intermediate manufacturing and ensures a more robust supply continuity for downstream API producers.
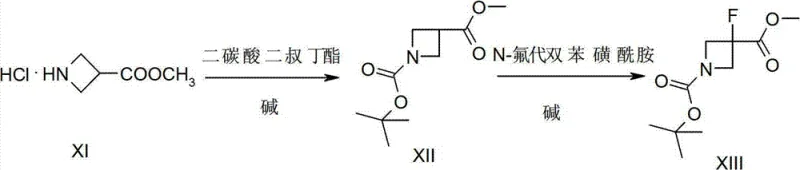
Mechanistic Insights into Electrophilic Fluorination of Azetidines
The core innovation of this technology lies in the precise execution of the fluorination step, which transforms the protected azetidine precursor into the desired 3-fluoro derivative. The mechanism relies on the generation of a stabilized enolate or carbanion at the 3-position of the azetidine ring using a strong, non-nucleophilic base such as lithium hexamethyldisilazide (LiHMDS) or sodium hexamethyldisilazide (NaHMDS). This deprotonation must be conducted under strictly controlled cryogenic conditions, typically between -70°C and -90°C, to prevent ring opening or decomposition of the strained four-membered system. Once the reactive anionic species is formed, it reacts with N-fluorobenzenesulfonimide (NFSI), acting as an electrophilic fluorine source. This specific reagent choice is critical because it delivers the fluorine atom with high regioselectivity and minimizes side reactions that are common with other fluorinating agents. The result is a clean transformation that preserves the stereochemical integrity and structural stability of the azetidine core.
Following the fluorination, the final step involves the hydrolysis of the methyl ester to the free carboxylic acid. This is achieved under mild alkaline conditions using sodium hydroxide in a methanol-water solvent system. The simplicity of this hydrolysis is a key factor in the process's overall success; it proceeds rapidly at ambient temperatures (around 20°C) and requires only a standard acid workup to isolate the product. From an impurity control perspective, the avoidance of transition metals means there is no risk of heavy metal contamination, a critical quality attribute for pharmaceutical intermediates. Additionally, the use of Boc protection early in the sequence ensures that the nitrogen atom remains inert throughout the fluorination and hydrolysis steps, preventing unwanted N-fluorination or polymerization side products. This mechanistic elegance ensures that the final product meets stringent purity specifications, often exceeding 98% purity without the need for complex chromatographic purification.
How to Synthesize 1-Boc-3-fluoroazetidine-3-carboxylic acid Efficiently
Implementing this synthesis in a GMP environment requires careful attention to temperature control and reagent stoichiometry, particularly during the fluorination stage. The process is designed to be operationally simple, utilizing common organic solvents like ethyl acetate, THF, and methanol, which facilitates easy solvent recovery and waste management. The following guide outlines the standardized protocol derived from the patent examples, ensuring reproducibility and safety. For detailed standard operating procedures and specific batch records, please refer to the technical guide below.
- Protect azetidine-3-carboxylic acid methyl ester with di-tert-butyl dicarbonate under alkaline conditions to form the Boc-protected intermediate.
- Perform electrophilic fluorination using N-fluorobenzenesulfonimide (NFSI) and a strong base like LiHMDS at low temperatures (-70°C to -90°C).
- Hydrolyze the methyl ester group under alkaline conditions using sodium hydroxide to obtain the final carboxylic acid derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthesis route offers transformative benefits that extend far beyond simple yield improvements. By shifting from an eight-step linear synthesis to a concise three-step convergent route, manufacturers can significantly reduce the lead time for high-purity pharmaceutical intermediates. The elimination of multiple isolation and purification stages reduces the consumption of raw materials, solvents, and energy, leading to substantial cost savings in the overall manufacturing budget. Furthermore, the removal of expensive and regulated transition metal catalysts simplifies the regulatory filing process and reduces the environmental footprint of the production facility. These factors combine to create a supply chain that is not only more cost-effective but also more resilient to market fluctuations and raw material shortages.
- Cost Reduction in Manufacturing: The primary driver of cost efficiency in this new method is the drastic reduction in unit operations. By cutting the number of reaction steps from eight to three, the labor hours, equipment occupancy time, and utility consumption are effectively slashed. Additionally, the replacement of costly reagents like Ruthenium catalysts and N-bromosuccinimide with more economical alternatives like NFSI and Boc anhydride directly lowers the bill of materials. The high yield of each individual step means that less starting material is wasted, further enhancing the economic viability of the process. This lean manufacturing approach allows suppliers to offer competitive pricing without compromising on quality, providing a clear financial advantage to downstream API manufacturers.
- Enhanced Supply Chain Reliability: Supply continuity is often threatened by complex syntheses that have multiple points of failure. The simplified three-step route minimizes these risks by reducing the number of potential bottlenecks. The starting material, azetidine-3-carboxylic acid methyl ester, is commercially available and stable, ensuring a consistent feedstock supply. Moreover, the reaction conditions are mild and do not require specialized high-pressure or high-temperature equipment, meaning that production can be easily scaled across multiple manufacturing sites if necessary. This flexibility ensures that procurement teams can secure long-term contracts with confidence, knowing that the supplier has the capacity to meet demand spikes without significant lead time delays.
- Scalability and Environmental Compliance: Scaling chemical processes often introduces new challenges regarding heat transfer and mixing, but this methodology is inherently scalable due to its reliance on standard solution-phase chemistry. The exothermic nature of the fluorination step is easily managed at low temperatures, and the workup procedures involve simple liquid-liquid extractions that translate well from the laboratory to the pilot plant and eventually to commercial tonnage production. From an environmental standpoint, the process generates less hazardous waste compared to the heavy-metal-catalyzed alternatives. The absence of toxic metals simplifies wastewater treatment and disposal, aligning with modern green chemistry principles and helping pharmaceutical companies meet their sustainability goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of 3-fluoroazetidine derivatives. These answers are derived directly from the experimental data and technical specifications outlined in the patent literature, providing clarity on the feasibility and advantages of this specific synthetic route. Understanding these details is crucial for making informed sourcing decisions.
Q: What is the total yield advantage of this new synthesis method compared to conventional routes?
A: The novel method achieves a total yield of approximately 74% for the key intermediate and up to 99% for the final hydrolysis step, significantly outperforming conventional 8-step routes which often suffer from cumulative yield losses resulting in totals as low as 2.46%.
Q: Why is N-fluorobenzenesulfonimide (NFSI) preferred for the fluorination step?
A: NFSI allows for mild and selective electrophilic fluorination at the 3-position of the azetidine ring under controlled low-temperature conditions, avoiding the harsh reagents and transition metal catalysts required in older methodologies.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the process utilizes readily available starting materials, avoids expensive transition metals like Ruthenium, and employs simple workup procedures such as extraction and crystallization, making it highly amenable to kilogram-to-ton scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Fluoroazetidine Derivative Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the successful development of new medicines. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can support your project from early-stage clinical trials through to full-scale commercialization. We are committed to maintaining stringent purity specifications and operate rigorous QC labs to guarantee that every batch of 1-Boc-3-fluoroazetidine-3-carboxylic acid meets the highest industry standards. Our state-of-the-art facilities are equipped to handle the low-temperature fluorination chemistry required by this patent, delivering consistent quality and reliability.
We invite you to collaborate with us to optimize your supply chain and accelerate your drug development timelines. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our advanced manufacturing capabilities can add value to your organization. Let us be your partner in bringing innovative therapies to market faster and more efficiently.